NEONATOLOGY: MANAGEMENT, PROCEDURES, ON-CALL PROBLEMS, DISEASES, AND DRUGS - part 8 ppsx
Bạn đang xem bản rút gọn của tài liệu. Xem và tải ngay bản đầy đủ của tài liệu tại đây (386.9 KB, 95 trang )
3. Consumption of high-dose folic acid must be under a physician's supervision because there
is still little information regarding its long-term effects and symptoms of pernicious anemia may be
obscured, with the potential to result in serious neurologic damage.
4. Because 4 mg of folic acid did not prevent all NTDs in the MRC study, patients should be
cautioned that folic acid supplementation does not preclude the need for counseling or consideration
of prenatal testing for NTDs.
C. The American Academy of Pediatrics Committee on Genetics has endorsed the U.S.
Department of Health and Human Services (1992) recommendation, as follows:
1. All women of childbearing age (15-44 years) in the United States who are capable of
becoming pregnant should consume 0.4 mg of folic acid per day for the purpose of reducing their
risk of having a pregnancy affected by spina bifida or other NTDs. This amount of folic acid is
estimated to reduce the NTD risk by 50-70%. This amount (0.4 mg) is also the US Recommended
Daily Allowance of folic acid.
2. An intake of >1 mg is not generally recommended.
3. Folic acid should ideally be taken at least 1 month before conception and at least through the
first month of gestation.
D. Sources of folic acid
1. Dietary. The average diet in the United States contains 0.2 mg of folate, which is less
bioavailable than folic acid. Folate intake of 0.4 mg/day can be achieved through careful selection of
folate-rich foods (spinach and other leafy green vegetables, dried beans, peas, liver, and citrus fruits).
Some breakfast cereals are fortified with folic acid. Since January 1998, enriched foods (including
flour, cornmeal, pasta, and rice) are fortified in folic acid by order of the US Food and Drug
Administration.
2. Supplementation. Folic acid is available over the counter in dosages up to 0.8 mg. Folic acid
is also available by prescription in 1-mg tablets. Prenatal vitamins contain 0.8 or 1 mg of folic acid. A
survey by the March of Dimes revealed that only 27% of nonpregnant women 18-45 years of age
took a vitamin preparation containing folic acid in 2001. Awareness of the U.S. Public Health Service
recommendation regarding folic acid did more than double from 1995 to 2002 (from 15 to 32%) for
the same group. Multiple sources are available to provide educational material to the public (March
of Dimes: 1-888-MODIMES or
, , .
org, ).
E. Current epidemiologic and biochemical evidence suggests that NTDs are not primarily due to
folate insufficiency but rather arise from changes in the metabolism of folate and possibly B
12
in
predisposed women. The mechanisms may also involve homocysteine metabolism. Polymorphisms
of methylene tetrahydrofolate reductase and other genes encoding proteins involved in folate
metabolism may be associated with an increased frequency of NTDs. Of further interest is that the
homocysteine-lowering effect of folic acid supplementation may also reduce the risk for
cardiovascular disease.
F. Intestinal hydrolysis of dietary folate is not impaired in mothers who have had infants with
NTDs, although the response curve to a folate-enriched meal appears to differ significantly from that
of mothers who have not had infants with NTDs.
V. Prenatal detection of NTDs
A. Prenatal screen using maternal serum AFP at 14-16 weeks' gestation. Elevated levels (>2.5
multiples of the mean, which are adjusted to gestational age) are indicative of open NTDs at a
sensitivity of 90-100%, a specificity of 96%, and a negative predictive value of 99-100% but a low
positive predictive value.
B. Prenatal diagnosis. Documentation of an elevated maternal serum AFP is followed by:
1. Genetic counseling. The physician needs to make sure that the patient receives information
regarding her risk for NTDs and other conditions with elevated AFP (gastroschisis or other
conditions leading to fetal skin defects), to evaluate causes of possible false-positive results
(imprecise dates or twin pregnancies), to learn about options regarding further evaluation (see later
discussion), and to provide nondirective counseling regarding treatment options.
2. Detailed fetal ultrasonography with anomaly screening. In skilled hands, a detailed
ultrasonogram can be extremely sensitive and specific for detection of NTDs. Sonographic
determination of the level of the lesion has been shown to be useful in predicting the ambulatory
potential of fetuses with NTDs. Ultrasonography is also done to rule out other major congenital
defects.
3. Measurement of the amniotic fluid AFP and acetylcholinesterase. Amniocentesis is
usually done between 16 and 18 weeks' gestation, although it can technically be done as early as 14
weeks' gestation. If indicated, karyotype can also be obtained. The detection rate for anencephaly and
open spina bifida is 100% when results of amniotic fluid acetylcholinesterase and AFP are combined,
with a false-positive rate of only 0.04%.
VI. Management: anencephaly
A. Approximately 75% are stillborn, and most live-born infants with anencephaly die within the
first 2 weeks of birth.
B. Considering the 100% lethality of anencephaly, usually only supportive care is given: warmth,
comfort, and enteral nutrition. Support services for the family, including social work and genetic and
general counseling, are essential. There are some ethically controversial issues regarding the extent
of care and other issues (eg, organ donation), and it may be advisable to involve other support
systems (eg, ethics committees, support groups, or religious guidance [if desired by the family]).
VII. Management: encephalocele
A. Physical examination and initial management. In addition to the general principles of
neonatal resuscitation, an especially careful physical examination is indicated. Look for associated
malformations. As mentioned in Neural Tube Defects, section III,C, some genetic publications list
up to 50 syndromes associated with NTDs. We recommend that the child be given nothing by mouth
until the consultations by subspecialties such as neurosurgery and, if indicated, genetic tests are done
and the need for immediate treatment (perhaps surgery) is assessed. Imaging studies
(ultrasonography, CT, and MRI) should be arranged.
B. Neurosurgical intervention may be indicated to prevent ulceration and infection, except in
those cases with massive lesions and marked microcephaly. The encephalocele and its contents are
often excised because the brain tissue within is frequently infarcted and distorted. Surgery may be
deferred, depending on the size, skin coverage, and location. Ventriculoperitoneal shunt (VP)
placement may be required because as many as 50% of cases have secondary hydrocephalus.
C. Counseling and long-term outcome. A multidisciplinary approach is necessary to counsel the
family regarding recurrence risk, long-term outcome, and follow-up. The family should be informed
about the availability of support groups (March of Dimes and others; March of Dimes Birth Defect
Foundation can be reached at 1-888-MODIMES). The degree of developmental deficits is
determined mainly by the extent of herniation and location; cerebral hemispheres from both sides or
one side, the cerebellum, and even the brainstem can be involved. Visual deficits are common with
occipital encephaloceles. Motor and intellectual deficits are found in ~50% of patients.
VIII. Management: myelomeningocele. Although fetal surgery for NTDs remains controversial,
many maternal-fetal specialists believe that this option should be mentioned to parents. After birth, a
multidisciplinary team approach, including the primary care physician, geneticist, genetic counselor,
neonatologist, urologist, neurosurgeon, orthopedic surgeon, and social worker, is necessary.
A. Physical examination should include careful evaluation for other malformations (see section
VII,A). In addition, special efforts should be made to correlate motor, sensory, and sphincter function
and reflexes to the functional level of lesion (Table 72-2).
1. Extent of neurologic dysfunction correlates with the level of the spinal cord lesion.
2. Paraplegia below the level of the defect.
3. The presence of the anal wink and anal sphincter tone suggests functioning sacral spinal
segments and is prognostically important. In one study, 90% of patients with a positive anocutaneous
reflex were determined to be "dry" on a regimen of intermittent catheterization as opposed to 50% of
those with a negative reflex.
B. Initial management. In addition to following the general principles of neonatal resuscitation
and newborn care, appropriate management of the spinal lesion is essential.
1. There are institutional differences in the specifics of how to cover the lesion, and provision of
a sterile cover can be achieved by several means. Some surgeons do prefer to have only a sterile
plastic material or wrap applied to the lesion and ask to avoid contact with gauze or other material
that could adhere to the tissue and result in mechanical damage when removed. It is advisable to try
to keep the defective area moist while avoiding bacterial contamination. If tolerated, the patient
should be positioned on the side.
TABLE 72-1. CAUSES OF NEONATAL SEIZURES
Perinatal asphyxia
Intracranial hemorrhage
Subarachnoid hemorrhage
Periventricular or intraventricular hemorrhage
Subdural hemorrhage
Metabolic abnormalities
Hypoglycemia
Hypocalcemia
Electrolyte disturbances: hypo- and hypernatremia
Amino acid disorders
Congenital malformations
Infections
Meningitis
Encephalitis
Syphilis, cytomegalovirus infections,
toxoplasmosis
Cerebral abscess
Drug withdrawal
Toxin exposure (particular local anesthetics)
Inherited seizure disorders
Benign familial epilepsy
Tuberous sclerosis
Zellweger syndrome
Pyridoxine dependency
TABLE 72-2. CORRELATION AMONG LEVEL OF MYELOMENINGOCELE, LEVEL OF CUTANEOUS
SENSATION, SPHINCTER FUNCTION, REFLEXES, AND POTENTIAL FOR AMBULATION
Level of lesion Innervation
Cutaneous sensation
(pinprick)
Sphincter
function
Reflexes
Ambulation
potential
Thoracolumbar T12-L2
Groin (L1)
Anterior upper thigh (L2)
Full braces
Wheelchair bound
Lumbar L3-L4
Anterior lower thigh and
knee (L3)
Medial leg (L4)
Knee jerk
May ambulate with
braces and crutches
Lumbosacral L5-S1
Lateral leg and medial
foot (L5)
Sole of foot (S1)
Ankle jerk
May ambulate with
or without short leg
braces
Sacral S2-S4
Posterior leg and thigh
(S2)
Middle of buttock (S3)
Medial buttock (S4)
Bladder and
rectal function
Anal wink
May ambulate
without braces
Voluntary muscle movements are difficult to elicit in newborns with myelomeningocele and are, therefore, not helpful
during initial evaluation. Furthermore, motor examination may be distorted initially by reversible spinal cord dysfunction
above the level of the actual defect induced by exposure of the open cord.
2. Be aware that a high rate of latex allergies has been reported in patients with NTDs. In some
centers, all patients with myelodysplasia are, therefore, considered at risk for anaphylaxis and other
allergic complications, and latex avoidance is practiced as a preventive protocol. One study showed
that after 6 years of a latex-free environment the prevalence of latex sensitization fell from 26.7% to
4.5% of children with spina bifida.
3. In most centers, patients are started on antibiotics (ampicillin and gentamicin) and are given
nothing by mouth.
4. Arrange for imaging studies to evaluate for hydrocephalus or other malformations detected
or suspected on physical examination.
C. Surgical management. Usually, closure of the back lesion is done within 24 or 48 h to prevent
infection and further loss of function.
D. Hydrocephalus is common and often noncommunicative secondary to Arnold-Chiari
malformation of the foramen magnum and upper cervical canal (usually type II), with resultant
downward displacement of the medulla, pons, and cerebellum and obstruction of CSF flow.
1. The risk of hydrocephalus is 95% for infants with thoracolumbar, lumbar, and lumbosacral
lesions and 63% for those with occipital, cervical, thoracic, or sacral lesions.
2. In most cases, hydrocephalus is not evident until after closure of the myelomeningocele, and
placement of a VP shunt may be required at a later date.
3. Aggressive treatment with early VP shunt placement may improve cognitive function.
4. Serial ultrasound scans are necessary to monitor progression of hydrocephalus because
ventricular dilation may occur without rapid head growth or signs of increased ICP. The
hydrocephalus usually becomes clinically overt 2-3 weeks after birth.
5. Despite treatment of the myelomeningocele and hydrocephalus, ~50% of these infants may
still succumb to death from aspiration, laryngeal stridor, and apnea attributable to the hindbrain
anomaly.
E. Urinary tract dysfunction is one of the major causes of morbidity and mortality after the first
year of life.
1. More than 85% of myelomeningoceles located above S2 are associated with neurogenic
bladder dysfunction, with urinary incontinence and ureteral reflux. Poor bladder emptying
immediately after NTD closure may be temporary ("spinal shock"), and improvement of bladder
function may be observed up to 6 weeks after repair.
2. Without proper management, hydronephrosis develops with progressive scarring and
destruction of the kidneys. Many of these infants succumb to urosepsis.
3. Renal ultrasonography and a voiding cystourethrogram may identify patients who could
benefit from anticholinergic medication, clean and intermittent catheterization, prophylactic
antibiotics, or early surgical intervention of the urinary tract.
4. Other associated renal anomalies include renal agenesis, horseshoe kidney, and ureteral
duplications.
F. Orthopedic complications
1. The lower extremities lack innervation and become atrophied.
2. Deformities of the foot, knee, hip, and spine are common as a result of muscle imbalance,
abnormal in utero positioning, or teratologic factors.
3. Hip dislocation or subluxation is usually evident within the first year of life, especially in
patients with midlumbar myelomeningocele.
4. Treatment of orthopedic abnormalities be instituted as soon as there is sufficient healing of
the back wound.
5. Physical therapists assist with proper positioning of the extremities to minimize contractures
and to maximize function.
G. Outcome of aggressive therapy
1. The overall mortality rate is now <15% by 3-7 years of age. One study revealed a survival
rate of infants with spina bifida of 87.2% for the first year. In multivariable analysis, factors
associated with increased mortality were low birth weight and high lesions.
2. Infants with sacral lesions have essentially no mortality.
3. The outcome in regard to the highest potential for ambulation depends largely on the level of
the original lesion (see
Table 72-2) and is modified by the orthopedic treatment and complications
(see section VIII,F).
4. The majority of children with lumbar myelomeningocele score within the normal range on
intelligence and achievement tests, with the greatest and possibly progressive deficits on
performance IQ, arithmetic achievement, and visuomotor integration, while keeping pace on reading
and spelling.
5. An IQ >80 is found in essentially all patients with lesions below S1.
6. Approximately 50% of survivors with thoracolumbar lesions have IQ >80.
7. Cognitive function is improved in the presence of favorable socioeconomic and
environmental factors.
IX. Management: spina bifida occulta
A. Neonatal features. The presence of spina bifida occulta is suggested by overlying abnormal
collections of hair, hemangioma, pigmented macule, aplasia cutis congenita, skin tag, subcutaneous
mass, cutaneous dimples, or tracts.
B. If undetected in the neonatal period, clinical presentation later in infancy includes the
following:
1. Delay in development of sphincter control.
2. Delay in walking.
3. Development of a foot deformity.
4. Recurrent meningitis.
5. A sudden deterioration may represent vascular insufficiency produced by tension on a
tethered cord, angulation of the cord around fibrous or related structures, or cord compression from a
tumor or cyst.
C. Diagnosis
1. Ultrasonography is useful for screening.
2. MRI provides superior anatomic details. The advantages of MRI are that contrast is not
needed and the infants are not exposed to radiation.
D. Surgical correction may be necessary in the newborn period to avoid the onset of symptoms.
Surgical release of a tethered cord or decompression of the spinal cord within 48 h of sudden
deterioration may completely or partially reverse recently acquired deficits.
REFERENCES
Agarwal SK et al: Outcome analysis of vesicoureteral reflux in children with myelodysplasia. J Urol
1997;157:980.
Anderson GD et al: The effect of cesarean section on intraventricular hemorrhage in the preterm
infant.
Am J Obstet Gynecol 1992;166:1091.
Aziz K et al: Province-based study of neurologic disability of children weighing 500 through 1249
grams at birth in relation to neonatal cerebral ultrasound findings.
Pediatrics 1995;95:837.
Batton DG et al: Current gestational age-related incidence of major intraventricular hemorrhage.
J
Pediatr 1994;125:623.
Bender J: Parental occupation and neural tube defect-affected pregnancies among Mexican
Americans.
J Occup Environ Med 2002;44:650.
Bernes SM, Kaplan AM: Evolution of neonatal seizures.
Pediatr Clin North Am 1994;41:1069.
Biggio et al: Can prenatal ultrasound findings predict the ambulatory status in fetuses with open spina
bifida? Am J Obstet Gynecol 2001;185:1016.
Birmingham PK et al: Do latex precautions in children with myelodysplasia reduce intraoperative
allergic reactions?
J Pediatr Orthop 1996;16:799.
Bluml S et al: Differentiation between cortical atrophy and hydrocephalus using
1
H MRS. Magn
Reson Med 1997;37:395.
Bower C et al: Absorption of pteroylpolyglutamates in mothers of infants with neural tube defects.
Br
J Nutr 1993a;69:827.
Bower C et al: Maternal folate status and the risk for neural tube defects. The role of dietary folate.
Ann NY Acad Sci 1993b;678:146.
Brock DJH et al: Prenatal diagnosis of neural tube defects with monoclonal antibody specific for
acetylcholinesterase. Lancet 1985;21:5.
Centers for Disease Control and Prevention: Economic costs of birth defects and cerebral
palsyUnited States, 1992.
MMWR Morb Mortal Wkly Rep 1995;44:694.
Centers for Disease Control and Prevention: Knowledge and use of folic acid by women of
childbearing ageUnited States, 1997.
MMWR Morb Mortal Wkly Rep 1997;46:721.
Clark RH et al: Intraventricular hemorrhage and high-frequency ventilation: a meta-analysis of
prospective clinical trials.
Pediatrics 1996;98:1058.
Committee on Genetics: Folic acid for the prevention of neural tube defects.
Pediatrics 1993; 92:493.
Dansky LV et al: Mechanisms of teratogenesis: folic acid and antiepileptic therapy.
Neurology
1992;42(suppl 5):32.
Dimmick JE, Kalousek DK: Developmental Pathology of the Embryo & Fetus. Lippincott, 1992.
Donn SM et al: Prevention of intraventricular hemorrhage with phenobarbital therapy: Now what?
Pediatrics 1986;77:779.
Dykes FD et al: Intraventricular hemorrhage: a prospective evaluation of etiopathogenesis.
Pediatrics
1980;66:42.
Dykes FD et al: Posthemorrhagic hydrocephalus in high-risk preterm infants: natural history,
management and long-term outcome.
J Pediatr 1989;114:611.
Fohr IP et al: 5,10-Methylentetrahydrofolate reductase genotype determines the plasma homocysteine
lowering effect of supplementation with 5-methyltetrahydrofolate or folic acid in healthy young
women.
Am J Clin Nutr 2002;75:275.
Fraser RK et al: The unstable hip and mid-lumbar myelomeningocele. J Bone Joint Surg 1992;
74:143.
Garland JS et al: Effect of maternal glucocorticoid exposure on risk of severe intraventricular
hemorrhage in surfactant-treated preterm infants.
J Pediatr 1995;126:272.
Gilman JT et al: Rapid sequential phenobarbital treatment of neonatal seizures.
Pediatrics 1989;
83:674.
Glick PL et al: Management of ventriculomegaly in the fetus.
J Pediatr 1984;105:97.
Goh D, Minns RA: Intracranial pressure and cerebral arterial flow velocity indices in childhood
hydrocephalus: current review.
Child Nerv Syst 1995;11:392.
Green DW et al: Nucleated erythrocytes and intraventricular hemorrhage in preterm neonates.
Pediatrics 1995;96:475.
Greitz D et al: A new view on the CSF-circulation with the potential for pharmacological treatment
of childhood hydrocephalus.
Acta Paediatr 1997;86:125.
Hanlo PW et al: Relationship between anterior fontanelle pressure measurements and clinical signs in
infantile hydrocephalus.
Child Nerv Syst 1996;12:200.
Horbar JD: Prevention of periventricular-intraventricular hemorrhage. In Sinclair J, Bracken MB
(eds): Effective Care of the Newborn Infant. Oxford University Press, 1992.
Hudgins RJ et al: Natural history of fetal ventriculomegaly.
Pediatrics 1988;82:692.
Hudgins RJ et al: Treatment of intraventricular hemorrhage in the premature infant with urokinase.
Pediatr Neurosurg 1994;20:190.
Kaempf JW et al: Antenatal phenobarbital for the prevention of periventricular and intraventricular
hemorrhage: a double-blind, randomized, placebo-controlled, multihospital trial.
J Pediatr
1990;117:933.
Laurence KM et al: Double blind randomized controlled trial of folate treatment before conception to
prevent recurrence of neural tube defects.
BMJ 1981;282:1509.
Lazzara A et al: Clinical predictability of intraventricular hemorrhage in preterm infants.
Pediatrics
1980;65:30.
Lemire RJ: Neural tube defects: clinical correlations.
Clin Neurosurg 1983;30:165.
Lemire RJ et al: Neural tube defects.
JAMA 1988;259:558.
Leviton A, Gilles F: Ventriculomegaly, delayed myelination, white matter hypoplasia, and
"periventricular" leukomalacia: how are they related? Pediatr Neurol 1996;15:127.
Leviton A et al: Antenatal corticosteroids appear to reduce the risk of postnatal germinal matrix
hemorrhage in intubated low birth weight newborns.
Pediatrics 1993;91:1083.
Lott JW et al: Umbilical artery catheter blood sampling alters cerebral blood flow velocity in preterm
infants. J Perinatol 1996;15:341.
Main DM, Mennuti MT: Neural tube defects: issues in prenatal diagnosis and counseling.
Obstet
Gynecol 1986;67:1.
March of Dimes and the Gallop Organization: Folic Acid and the Prevention of Birth Defects. A
National Survey of Pre-pregnancy Awareness and Behavior Among Women of Childbearing Age
1995-2001. March of Dimes, 2001.
Massager N et al: Anterior fontanelle pressure monitoring for the evaluation of asymptomatic infants
with increased head growth rate.
Child Nerv Syst 1996;12:38.
McCullough DC, Balzer-Martin LA: Current prognosis in overt neonatal hydrocephalus.
J Neurosurg
1982;57:378.
Medical Research Council Vitamin Study Research Group: Prevention of neural tube defects: results
of the Medical Research Council Vitamin Study.
Lancet 1991;338:131.
Meeropol E et al: Allergic reaction to rubber in patients with myelodysplasia.
N Engl J Med
1990;323:1072.
Ment LR et al: Antenatal steroids, delivery mode, and intraventricular hemorrhage in preterm infants.
Am J Obstet Gynecol 1995;172:795.
Ment LR et al: Low-dose indomethacin and prevention of intraventricular hemorrhage: a multicenter
randomized trial.
Pediatrics 1994a;93:543.
Ment LR et al: Low-dose indomethacin therapy and extension of intraventricular hemorrhage: a
multicenter randomized trial.
J Pediatr 1994b;124:951.
Ment LR et al: Neurodevelopmental outcome at 36 months' corrected age of preterm infants in the
multicenter indomethacin intraventricular hemorrhage prevention trial.
Pediatrics 1996; 98:714.
Michejda M et al: Present status of intrauterine treatment of hydrocephalus and its future.
Am J
Obstet Gynecol 1986;155:873.
Mills JL, Raymond E: Effects of recent research on recommendations for periconceptional folate
supplement use.
Ann NY Acad Sci 1993;678:137.
Morrow JD, Wachs TD: Infants with myelomeningocele: visual recognition memory and
sensorimotor abilities. Dev Med Child Neurol 1992;34:488.
Myianthopoulos NC, Melnick M: Studies in neural tube defects: epidemiologic and etiologic aspects.
Am J Med Genet 1987;26:783.
National Center for Health Statistics: Trends in Spina Bifida and Anencephalus in the United States,
1991-2001. Available from
Nelson KB, Grether JK: Can magnesium sulfate reduce the risk of cerebral palsy in very low
birthweight infants?
Pediatrics 1995;95:263.
Nieto A et al: Efficacy of latex avoidance for primary prevention of latex sensitization in children
with spina bifida.
J Pediatr 2002;140:370.
Noetzel MJ: Myelomeningocele: current concepts of management.
Clin Perinatol 1989;16:311.
Papile LS et al: Incidence and evolution of the subependymal intraventricular hemorrhage: a study of
infants with weights less than 1500 g.
J Pediatr 1978;92:529.
Perlman JM et al: Bilateral cystic periventricular leukomalacia in the premature infant: associated
risk factors.
Pediatrics 1996;97:822.
Philip AGS et al: Intraventricular hemorrhage in preterm infants: declining incidence in the 1980s.
Pediatrics 1989;84:797.
Poland RL: Vitamin E for prevention of perinatal intracranial hemorrhage.
Pediatrics 1990;85: 865.
Rasmussen AG et al: A comparison of amniotic fluid alpha-fetoprotein and acetylcholinesterase in
the prenatal diagnosis of open neural tube defects and anterior abdominal wall defects.
Prenat Diagn
1993;13:93.
Recommendations for the use of folic acid to reduce the number of cases of spina bifida and other
neural tube defects.
MMWR Morb Mortal Wkly Rep 1992;41(RR-14):1.
Resch B et al: Neurodevelopmental outcome of hydrocephalus following intra-/periventricular
hemorrhage in preterm infants: short- and long-term results.
Child Nerv Syst 1996;12:27.
Robbin M et al: Elevated levels of amniotic fluid α-fetoprotein: sonographic evaluation.
Radiology
1993;188:165.
Rodgers WB et al: Surgery of the spine in myelodysplasia. Clin Orthop Rel Res 1997;338:1.
Salafia CM et al: Maternal, placental, and neonatal associations with early germinal matrix/
intraventricular hemorrhage in infants born before 32 weeks' gestation.
Am J Perinatol 1995;12:429.
Sanders et al: The anocutaneous reflex and urinary continence in children with myelomeningocele.
Br J Urol 2002;89:720.
Sandovnick AD et al: Use of genetic counseling services for neural tube defects. Am J Med Genet
1987;26:811.
Schorah CJ et al: Possible abnormalities of folate and vitamin B
12
metabolism associated with neural
tube defects.
Ann NY Acad Sci 1993;678:81.
Sgouros S et al: Long-term complications of hydrocephalus.
Pediatr Neurosurg 1995;23:127.
Shalak L, Perlman JM: Hemorrhagic-ischemic cerebral injury in the preterm infant: current concepts.
Clin Perinatol 2002;29:745.
Shankaran S et al: Antenatal phenobarbital therapy and neonatal outcome: I. Effect on intracranial
hemorrhage.
Pediatrics 1996a;97:644.
Shankaran S et al: Antenatal phenobarbital therapy and neonatal outcome: II. Neurodevelopmental
outcome at 36 months.
Pediatrics 1996b;97:649.
Shankaran S et al: The effect of antenatal phenobarbital therapy on neonatal intracranial hemorrhage
in preterm infants.
N Engl J Med 1997;337:466.
Shaw GM et al: Epidemiological characteristics of phenotypically distinct neural tube defects among
0.7 million California births, 1983-1987.
Teratology 1994;49:143.
Shaw GM et al: Maternal periconceptional vitamin use, genetic variation of infant reduced folate
carrier (A80G), and risk of spina bifida.
Am J Med Genet 2002;108:1.
Shaw GM et al: Risk of neural tube defect-affected pregnancies among obese women.
JAMA
1996;275:1093.
Smithells RW et al: Further experience of vitamin supplementation for the prevention of neural tube
defect recurrences.
Lancet 1983;1:1027.
Stafstrom CE: Neonatal seizures.
Pediatr Rev 1995;16:248.
Stoneking et al: Early evolution of bladder emptying after meningomyelocele closure.
Urology
2001;58:767.
U.S. Department of Health and Human Services: Recommendations for the use of folic acid to reduce
the number of cases of spina bifida and other neural tube defects.
MMWR Morb Mortal Wkly Rep
1992;41:1.
Varvarigou A et al: Early ibuprofen administration to prevent patent ductus arteriosus in premature
newborn infants.
JAMA 1996;275:539.
Verget RG et al: Primary prevention of neural tube defects with folic acid supplementation: Cuban
experience. Prenat Diagn 1990;10:149.
Verma U et al: Obstetric antecedents of intraventricular hemorrhage and periventricular leukomalacia
in the low-birth-weight neonate.
Am J Obstet Gynecol 1997;176:275.
Vintzileos AM et al: Congenital hydrocephalus: review and protocol for perinatal management.
Obstet Gynecol 1983;62:539.
Vohr B, Ment LR: Intraventricular hemorrhage in the preterm infant.
Early Hum Dev 1996;44:1.
Volpe JJ: Intraventricular hemorrhage and brain injury in the premature infant. Pediatr Clin North
Am 1989a;2:361.
Volpe JJ: Neonatal seizures: current concepts and revised classification.
Pediatrics 1989b;84: 422.
Volpe JJ: Neurology of the Newborn, 3rd ed. Saunders, 1995.
Volpe JJ: Neurology of the Newborn, 4th ed. Saunders, 2001.
Warkany J: Hydrocephalus. In Warkany J (ed): Congenital Malformations. Year Book, 1971.
Weekes EW et al: Nutrient levels in amniotic fluid from women with normal and neural tube defect
pregnancies.
Biol Neonate 1992;61:226.
Wells JT, Ment LR: Prevention of intraventricular hemorrhage in preterm infants.
Early Hum Dev
1995;42:209.
Whitaker AH et al: Neonatal cranial ultrasound abnormalities in low birth weight infants: relation to
cognitive outcomes at six years of age.
Pediatrics 1996;98:719.
Whitelaw A et al: Phase I study of intraventricular recombinant tissue plasminogen activator for
treatment of posthaemorrhagic hydrocephalus. Arch Dis Child 1996;75:F20.
Wills KE et al: Intelligence and achievement in children with myelomeningocele.
J Pediatr Psychol
1990;15:161.
Wong LY, Paulozzi LJ: Survival of infants with spina bifida: a population study, 1979-94.
Paediatr
Perinat Epidemiol 2001;15:374.
CHAPTER 73. Perinatal Asphyxia
MANAGEMENT OUTLINE
I. Definition.
A. Perinatal asphyxia (from the Greek term sphyzein meaning "a stopping of the pulse") is a
condition caused by a lack of oxygen in respired air, resulting in impending or actual cessation of
apparent life.
B. Perinatal asphyxia is a condition of impaired blood gas exchange that, if it persists, leads to
progressive hypoxemia and hypercapnia with a metabolic acidosis.
C. Essential characteristics defined jointly by the American Academy of Pediatrics (AAP) and
the American College of Obstetricians and Gynecologists (ACOG) should be present: (1) profound
metabolic or mixed acidemia (pH <7.00) on umbilical cord arterial blood sample, if obtained; (2)
persistence of an Apgar score of 0-3 for >5 min; (3) neurologic manifestations in the immediate
neonatal period to include seizures, hypotonia, coma, or hypoxic-ischemic encephalopathy (HIE);
and (4) evidence of multiorgan system dysfunction in the immediate neonatal period.
D. Biochemical indices. There is no specific blood test to diagnose perinatal asphyxia.
1. The normal umbilical arterial base excess is a negative 6 mEq/L with -10 to -12 mEq/L as the
upper statistical limit of normal. Base excess > -20 mEq/L is required to show neurologic damage
associated with metabolic acidosis.
2. The precise value that is required to define damaging acidemia is not known. A pH <7.0
realistically represents clinically significant acidosis. Acidemia alone does not establish that hypoxic
injury has occurred.
E. Apgar score
1. Conceived to report on the state of the newborn and effectiveness of resuscitation. It is a poor
tool for assessing asphyxia. Low Apgar scores are unlikely to be the cause of morbidity but rather the
results of prior causes.
2. An infant with an Apgar score of 0-3 at 5 min, improving to ≥4 by 10 min, has >99% chance
of not having cerebral palsy (CP) at 7 years of age; 75% of children who develop CP have normal
Apgar scores at birth.
3. A 1996 revised AAP/ACOG statement again emphasized that the Apgar score alone should
not be used as evidence that neurologic damage was caused by hypoxia resulting in neurologic injury
or by inappropriate intrapartum management.
II. Incidence of asphyxia and its relationship to CP. The incidence of HIE is 2-9 in 1000 live term
births. The incidence of CP has not fallen despite improved obstetric and neonatal interventions and
remains at 1-2 in 1000 live term births. Only 8-17% of CP in term infants is associated with adverse
perinatal events suggestive of asphyxia; the cause of ≥90% of cases remains unknown. One cannot
state with a reasonable degree of medical certainty that CP in a given child was due to
intrapartum asphyxia merely because the physician can find no other explanation. The death
rate in term infants with HIE is ~11% and ~0.3 in 1000 live term births are severely affected. The
incidence of HIE, deaths, and handicap rates are all significantly higher for premature infants.
III. Mechanisms of asphyxia during labor, delivery, and the immediate postpartum period.
A. Interruption of the umbilical circulation (cord compression).
B. Inadequate perfusion of the maternal side of the placenta (maternal hypotension,
hypertension, abnormal uterine contractions).
C. Impaired maternal oxygenation (cardiopulmonary disease, anemia).
D. Altered placental gas exchange (placental abruption, previa, insufficiency).
E. Failure of the neonate to accomplish lung inflation and successful transition from fetal to
neonatal cardiopulmonary circulation.
IV. Pathophysiology.
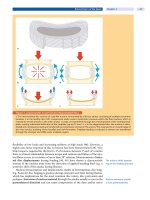
Figure 73-1 shows the corresponding respiratory and cardiovascular effects
during prolonged asphyxia.
A. Adaptive responses of the fetus or newborn to asphyxia. The fetus and neonate are much
more resistant to asphyxia than adults. In response to asphyxia, the mature fetus redistributes the
blood flow to the heart, brain, and adrenals to ensure adequate oxygen and substrate delivery to these
vital organs.
B. Impairment of cerebrovascular autoregulation results from direct cellular injury and cellular
necrosis from prolonged acidosis and hypercarbia.
C. The majority of neuronal disintegration occurs after termination of the asphyxial insult
because of persistence of abnormal energy metabolism and low adenosine triphosphate (ATP) levels.
A cascade of deleterious events is triggered, resulting in formation of free radicals, increased
extracellular glutamate, increased cytosolic Ca
2+
, and delayed cell death.
1. Effects of increased cytosolic Ca
2+
a. Degradation of cellular lipids, proteins, and DNA via activation of phospholipases,
proteases, and nucleases.
b. Uncoupling of oxidative phosphorylation.
c. Increased release of glutamate.
d. Production of free radicals as the result of oxygenation of arachidonic acid and
hypoxanthine and accumulation of nitric oxide via activation of nitric oxide synthetase.
2. Effects of increased extracellular glutamate. Immediate neuronal death (in minutes) as a
FIGURE 73-1. Respiratory and cardiovascular effects during prolonged asphyxia.
result of osmolar lysis from influx of Na
+
, Cl
-
, and H
2
O; delayed neuronal death (in hours) from
activation of glutamate receptors, Ca
2+
influx, and the effects of increased cytosolic Ca
2+
.
D. Major circulatory changes during asphyxia
1. Loss of cerebrovascular autoregulation under conditions of hypercapnia, hypoxemia, or
acidosis. Cerebral blood flow (CBF) becomes "pressure passive," leaving the infant at risk for
cerebral ischemia with systemic hypotension and cerebral hemorrhage with systemic hypertension.
2. Increase in CBF secondary to redistribution of cardiac output, initial systemic hypertension,
loss of cerebrovascular autoregulation, and local accumulation of vasodilator factors (H
+
, K
+
,
adenosine, and prostaglandins).
3. With prolonged asphyxia, there is a decrease in cardiac output, hypotension, and a
corresponding fall in CBF. In general, brain injury occurs only when the asphyxia is severe enough to
impair CBF.
E. The postasphyxial human newborn is in a persistent state of vasoparalysis and cerebral
hyperemia, the severity of which is correlated with the severity of the asphyxial insult.
Cerebrovascular hemorrhage may occur on reperfusion of the ischemic areas of the brain. However,
when there has been prolonged and severe asphyxia, local tissue recirculation may not be restored
because of collapsed capillaries in the presence of severe cytotoxic edema.
F. Cerebral edema is a consequence of extensive cerebral necrosis rather than a cause of ischemic
cerebral injury.
G. Regional vulnerability changes with postconceptional age (PCA) and as the infant matures.
1. Periventricular white matter is most severely affected in infants <34 weeks' PCA. The
"watershed" areas between the anterior and middle cerebral arteries and between the middle and
posterior cerebral arteries are predominantly involved in term infants.
2. Areas of brain injury in profound asphyxia correlate temporally and topographically with the
progression of myelinization and of metabolic activity within the brain at the time of the injury.
White matter is, therefore, more susceptible to hypoxic injury.
3. The topography of brain injury observed in vivo corresponds closely to the topography of
glutamate receptors.
4. When CBF is increased in response to asphyxia, regional differences exist such that there is
relatively more blood flow to the brainstem than to higher cerebral structures.
V. Neuropathologic findings
A. Cortical changes. Cortical edema, with flattening of cerebral convolutions, is followed by
cortical necrosis until finally a healing phase results in gradual cortical atrophy. Cortical atrophy, if
severe, may result in microcephaly.
B. Selective neuronal necrosis is the most common type of injury observed in neonatal HIE.
C. Other findings seen in term infants include status marmoratus of the basal ganglia and
thalamus (the marbled appearance is a result of the characteristic feature of hypermyelinization) and
parasagittal cerebral injury (bilateral and usually symmetric, with the parieto-occipital regions
affected more often than those regions anteriorly).
D. Periventricular leukomalacia (PVL) is hypoxic-ischemic necrosis of periventricular white
matter resulting from cerebral hypoperfusion and the vulnerability of the oligodendrocyte within the
white matter to free radicals, excitotoxin neurotransmitters, and cytokines. Injury to the
periventricular white matter is the most significant problem contributing to long-term neurologic
deficit in the premature infant, although it does occur in sick full-term infants as well. The incidence
of PVL increases with the length of survival and the severity of postnatal cardiorespiratory
disturbances. PVL involving the pyramidal tracts usually results in spastic diplegic or quadriplegic
CP. Visuoperception deficits may result from involvement of the optic radiation.
E. Porencephaly, hydrocephalus, hydranencephaly, and multicystic encephalomalacia may
follow focal and multifocal ischemic cortical necrosis, PVL, or intraparenchymal hemorrhage.
F. Brainstem damage is seen in the most severe cases of hypoxic-ischemic brain injury and
results in permanent respiratory impairment.
VI. Clinical presentation
A. The majority of infants who experience intrauterine hypoxic-ischemic insults do not exhibit
overt neonatal neurologic features or subsequent neurologic evidence of brain injury. It is
generally accepted that after acute perinatal asphyxia there should be an acute encephalopathy, often
accompanied by multiorgan malfunction.
B. Occurrence of neonatal neurologic syndrome shortly after birth is a sine qua non for recent
(ie, intrapartum) insult. Prenatal insult may also have occurred. The primary signs of central nervous
system (CNS) injury in the term infant include seizures, abnormal respiratory patterns (apnea),
posturing and movement disorders, impaired suck, and jitteriness. The absence of this neonatal
neurologic syndrome rules out intrapartum insult as the cause of major brain injury.
C. The severity of HIE correlates with the duration and severity of the asphyxial insult. A
constellation of neurologic signs evolves over the first 72 h of life best characterized by Sarnat and
Sarnat in 1976: stage I (hyperalert, awake state), stage 2 (lethargic, obtunded, hypotonic, seizures),
and stage 3 (stuporous, comatose, flaccid, posturing). Moderately to severely affected infants are
usually obtunded if not comatose, with generalized hypotonia and paucity of spontaneous
movements. Depressed reflexes and cranial nerve palsies are common findings. Presentation of
hypertonicity and irritability generally are not noted until the second week of life.
D. Occurrence of seizures within the first 12-24 h after birth is indicative of intrapartum insult
until proven otherwise. Seizures may also be secondary to hypoglycemia. Perlman and Risser (1996)
showed that the combination of a 5-min Apgar score of ≤5 and the need for intubation in the delivery
room in association with an umbilical cord arterial pH ≤7.00 has an odds ratio of 340 for the
development of seizures in the first 24 h of life.
E. Hypoxic-ischemic spinal cord injury. Ischemic injury to anterior horn cells within the spinal
cord gray matter is relatively common among hypotonic and hyporeflexic neonates after severe
perinatal hypoxia-ischemia. Electromyographic examinations show injury to the lower motor neuron
above the level of the dorsal root ganglion (Clancy et al, 1989).
F. Clinical presentation may be further obscured by the coexistence of skull fracture, subdural
hematoma, or subarachnoid hemorrhage resulting from traumatic delivery.
G. Multiple organ involvement. A prospective study by Martin-Ancel et al (1995) showed that
involvement of 1 or more organs occurred in 82% of infants with perinatal asphyxia. The central
nervous system (CNS) was the organ most frequently involved (72%). Severe CNS injury always
occurred with involvement of other organs, although moderate CNS involvement was isolated in 20%
of the infants. Renal involvement occurred in 42% of the infants, pulmonary involvement in 26%,
cardiac involvement in 29%, and gastrointestinal involvement in 29%. Fifteen percent of neonates
experienced renal failure, and 19% had respiratory failure. All of the infants in this study with an
Apgar score <5 at 5 min had severe involvement of at least 1 organ, whereas 90% of the infants with
an Apgar score ≥5 at 5 min did not have severe involvement of any organ.
1. Cardiovascular system. Shock, hypotension, tricuspid insufficiency, myocardial necrosis,
congestive heart failure, and ventricular dysfunction.
2. Renal function. Oliguria-anuria, acute tubular or cortical necrosis (hematuria, proteinuria),
and renal failure.
3. Hepatic function. Elevated serum γ-glutamyl transpeptidase activity, ammonia and indirect
bilirubin, and decreased clotting factors at 3-4 days' postnatal age in moderate to severe asphyxia.
4. Gastrointestinal tract. Paralytic ileus or delayed (5-7 days) necrotizing enterocolitis.
5. Lungs. Respiratory distress syndrome (see
Chapter 74) from surfactant deficiency or
dysfunction, pulmonary hemorrhage (shock lung), and persistent pulmonary hypertension (see
Chapter 62).
6. Hematologic system. Thrombocytopenia can result from shortened platelet survival or
disseminated intravascular coagulopathy. Increased numbers of nucleated red blood cells have been
reported (see later discussion).
7. Metabolic. Acidosis, hypoglycemia (hyperinsulinism), hypocalcemia (increased phosphate
load, correction of metabolic acidosis), and hyponatremia/syndrome of inappropriate antidiuretic
hormone secretion (SIADH).
8. Acute Perinatal Asphyxia Scoring System. A simple scoring system can be used to identify
those newborns depressed at birth who are at greatest risk for multiple organ system sequelae. The
scoring system is composed of the 5-min Apgar, umbilical artery base deficit, and fetal heart rate
(FHR) monitor tracing (Carter et al, 1998). Multiple organ system morbidity was more likely to occur
when the score exceeds 6.
VII. Diagnosis. Recognition of neonatal HIE depends principally on information gained from a
careful history and a thorough physical examination with appropriate laboratory studies as outlined
previously. Neurodiagnostic and neuroimaging studies can help determine the extent of the injury and
may also be of value prognostically.
A. Antenatal indicators of uteroplacental insufficiency or fetal compromise (see also Chapter
1) may include the following:
1. Reactive FHR and subsequent prolonged FHR deceleration suggestive of a sudden
catastrophic event (pattern of acute asphyxia).
2. Reactive FHR, which, during labor, becomes nonreactive, associated with rising FHR
baseline and repetitive late decelerations (pattern of intrapartum asphyxia).
3. A persistent nonreactive FHR tracing with a fixed baseline rate, from admit until
delivery, is suggestive of prior neurologic injury. This FHR pattern is often associated with reduced
fetal movement, old passage of meconium, oligohydramnios, and abnormal fetal pulmonary
vasculature (persistent pulmonary hypertension).
4. FHR patterns are not always specific, with a substantial false-positive rate. Improving the
predictive value of FHR pattern in detecting intrapartum asphyxia may require supplementary tests:
a. Fetal vibroacoustic stimulation
b. Fetal pulse oximetry
c. A decreased biophysical profile score
d. An amniotic fluid index ≤5.
e. An increased pulsatility index in the umbilical artery or decreased fetal cerebral resistance
on Doppler ultrasonography.
5. ACOG cautions against using terms such as asphyxia, hypoxia, and fetal distress when
applied to continuous electronic fetal monitoring or auscultation.
B. EEG. Evolution of EEG changes may provide information on the severity of the asphyxial
injury, and the type of EEG abnormality may be indicative of a specific pathologic variety.
Identification of EEG abnormalities within the first hours after delivery may be helpful in selecting
infants for treatment with neuroprotective agents.
C. Computed tomography (CT) scan. The value of CT in the assessment of diffuse cortical
neuronal injury is most apparent several weeks after severe asphyxial insults. It is of particular value
in the identification of focal and multiple ischemic brain injury. During the first week after an insult,
the striking, bilateral, diffuse hypodensity reflects marked cortical neuronal injury, with associated
edema corresponding closely to the occurrence of maximum intracranial pressure.
D. Ultrasonography is the method of choice for routine screening of the premature brain. It is
of major value in the identification of intraventricular hemorrhage and necrosis of basal ganglia and
thalamus. It is superior to CT in identifying both the acute and subacute-chronic manifestations of
periventricular white matter injury. Its limitations in the first weeks of life include its inability to
reliably identify mild injury, to visualize lesions that are peripherally located, and to distinguish
between hemorrhagic and ischemic lesions in the cerebral parenchyma.
E. Magnetic resonance imaging (MRI) is the technique of choice for evaluation of hypoxic-
ischemic cerebral injury in term and premature newborns. The advantages of MRI include the
following:
1. It does not expose the neonate to radiation.
2. It demonstrates better anatomic imaging detail and resolution than CT, especially of the deep
cortical structures (eg, the basal ganglia and thalamus) and corticospinal tracts.
3. It clearly demonstrates the myelinization delay that almost invariably accompanies asphyxial
brain injury. MRI may provide insight into the timing and duration of the asphyxial injury. Delayed
myelinization is a negative predictor of long-term neurodevelopmental outcome.
4. MRI is probably the best method available to diagnose hypoxic brain injuries in mildly to
moderately affected patients and to detect discrete lesions of the cerebellum and brainstem.
5. It may provide clues to other disorders (eg, metabolic or neurodegenerative disorders) that
may also present as obtundation or coma in the newborn period.
6. In experienced hands, ischemic lesions can be identified as early as 24 h after the insult.
7. MRI can help differentiate between partial asphyxia and anoxia.
a. Partial asphyxia. Injury is caused primarily by mild or moderate hypoxia or hypotension.
Regions of the brain with the most tenuous perfusion are affected, and susceptibility varies as the
infant matures (ie, periventricular white matter in premature infants and "watershed" areas in term
infants). Deep gray matter structures of the cerebrum are typically spared.
b. Anoxia. Injury is the result of a cardiorespiratory arrest or profound hypotension. The
volume of damaged brain varies with the duration of the injury. An arrest of long duration (≥25 min)
damages nearly the entire brain. Arrests of shorter duration show specific patterns that vary with
PCA: at 26-32 weeks, the lateral thalami are primarily affected; at 34-36 weeks, the lentiform nucleus
and hippocampus and the perirolandic cortex are affected; and by 40 weeks, the corticospinal tracts
from the internal capsule to the perirolandic cortex are affected. More severe or prolonged events
result in injury to the optic radiations.
8. MRI demonstrates the structural sequelae of asphyxial injury on follow-up and has prognostic
value. Repeat MRI at 3 months of age will usually show the full extent of brain injury.
F. Evoked electrical potentials (auditory, visual, or somatosensory) performed within the first
hours of life may help to select infants for treatment with neuroprotective agents. They also have
prognostic value in defining areas of CNS damage. Persistence of deficits beyond the neonatal period
correlates with persistence of other signs of brain injury.
G. Potentially useful techniques
1. Magnetic resonance spectroscopy (MRS) provides a measure of "energy reserve." Using
phosphorus-/(
31
P) MRS, it has been shown that asphyxiated newborns tend to have lower
phosphocreatine/inorganic phosphate ratios (impaired brain oxidative phosphorylation) and lower
ATP/total phosphorus ratios than normal patients.
2. Proton MRS allows noninvasive observations to be made of the derangement of cerebral
metabolites (N-acetylaspartate (NAA) and lactic acid) when oxidative phosphorylation is impaired.
The normalization of phosphorous metabolite ratios with time may reflect loss of severely affected
neurons. Neuronal loss, gliosis, and delay in myelination would be reflected by a relative loss of
NAA.
3. Near-infrared spectroscopy on the first day after injury may demonstrate increased cerebral
venous oxygen saturation and decreased cerebral oxygen extraction, despite increased cerebral
oxygen delivery, suggestive of a postasphyxial decrease in oxygen utilization.
VIII. Management
A. Optimal management is prevention. The first goal is to identify the fetus being subjected to or
likely to experience hypoxic-ischemic insults with labor and delivery.
B. Immediate resuscitation. Any newborn that is apneic at birth must be promptly resuscitated
because it cannot be determined whether the infant is in primary or secondary apnea.
1. Maintenance of adequate ventilation. Use an assisted ventilatory rate to maintain
physiologic levels of PCO
2
. Hypercarbia can further increase cerebral intracellular acidosis and
impair cerebrovascular autoregulation, whereas hypocarbia (PaCO
2
<20-25 mm Hg) has been
associated with PVL in preterm infants and late-onset sensorineural hearing loss in full-term infants.
2. Maintenance of adequate oxygenation (PaO
2
>40 in premature infants and PaO
2
>50 in
term infants). Avoid hyperoxia (see later discussion), which may lead to additional brain injury from
possible reduction in CBF and vaso-obliterative changes.
3. Maintenance of adequate perfusion. Maintain arterial blood pressure in the "normal" range
for gestational age and weight. Volume expanders and inotropic support are often required. With the
loss of cerebrovascular autoregulation, it is important to avoid systemic hypotension and
hypertension.
4. Correct metabolic acidosis with cautious use of volume expanders. The primary objective is
to sustain tissue perfusion. Perfuse or lose! Use bicarbonate only when cardiopulmonary resuscitation
(CPR) is prolonged and the infant remains unresponsive. Bicarbonate administration may lead to
hypercarbia and intracellular acidosis and increase lactate.
5. Maintain a normal serum glucose level (~75-100 mg/dL) to provide adequate substrate for
brain metabolism. Avoid hyperglycemia to prevent hyperosmolality and a possible increase in brain
lactate levels.
6. Control of seizures
a. Phenobarbital is the drug of choice. It is usually continued until the EEG is normal and
there are no clinical seizures for ≥2 months. The benefit of prophylactic therapy remains
controversial. High-dose phenobarbital (40 mg/kg) reduced the incidence of seizures and improved
neurologic outcome at 3 years in term asphyxiated newborns (Hall et al, 1998).
b. If seizures persist despite therapeutic phenobarbital levels, diazepam, lorazepam, and
phenytoin may be used (for dosages and other pharmacologic information, see
Chapter 80).
7. Prevention of cerebral edema. The cornerstone of prevention of serious brain swelling is
avoidance of fluid overload. Maintain slight to moderate fluid restriction (eg, 60 mL/kg). If cerebral
edema is severe, further restriction of fluid intake to 50 mL/kg is imposed. Observe the infant for
SIADH. Glucocorticoids and osmotic agents are not recommended.
C. Potential new therapies should aim at preventing delayed neuronal death once an
asphyxial insult has occurred. It is estimated that there is a 6- to 12-h window of opportunity after
acute asphyxia whereby administration of a neuroprotective agent could reduce or prevent brain
damage. Protecting the brain from injury would depend on the baseline fetal brain status.
1. Magnesium has an inhibitory effect on excitation of the N-methyl-D- aspartate type of
glutamate receptors and competitively blocks Ca
2+
entry through voltage-dependent Ca
2+
channels
during hypoxia. Apnea may occur, and higher doses carry a significant risk of hypotension. Use of
magnesium sulfate (MgSO
4
) remains controversial.
2. Prevention of free radical formation
a. Xanthine oxidase inhibitor. In a pilot study (Van Bel et al, 1998), allopurinol reduced free
radical formation and enhanced electrical brain activity in severely asphyxiated newborns. In
addition, allopurinol reduced nonprotein iron (a prooxidant).
b. Resuscitation with room air. In the Resair 2 trial (Saugstad 2001), room air-resuscitated
infants recovered more quickly as assessed by time to first cry, 5-min Apgar score, and sustained
pattern of respiration. Neonates resuscitated with 100% oxygen manifest biochemical changes
indicative of prolonged oxidative stress at 4 weeks of age (Vento et al, 2001).
3. Excitatory amino acid antagonists.
4. Calcium channel blockers.
5. Inhibition of nitric oxide production. Increased plasma nitric oxide levels has been shown
as a marker for severity of brain injury and poor neurologic outcome (Shi et al, 2000).
6. Selective head cooling. Hypothermia is thought to protect the brain from injury by preventing
the decline in high-energy phosphates. Phosphocreatine and adenosine triphosphate are maintained
while cerebral lactate levels are reduced. Selective head cooling coupled with mild systemic
hypothermia was found to be safe in a group of asphyxiated term infants (Gunn et al, 1998).
7. Any multicentered trial testing a new therapy to prevent or limit brain injury will require early
enrollment soon after birth in infants at greatest risk of developing the sequelae of HIE.
IX. Prognosis. Most survivors of perinatal asphyxia do not have major sequelae. Peliowski and
Finer (1992) showed that the overall risk of death for children with all stages of HIE combined was
12.5%, 14.3% for neurologic handicap, and 25% for death plus handicap. Depressed FHR, meconium-
stained amniotic fluid, low "extended" Apgar scores, low scalp and cord pH, or clinical signs of
neurologic depression soon after birth signify the acute clinical condition of the newborn. However,
their predictive value for later neurodevelopmental outcome is less than satisfactory, especially when
taken individually. Furthermore, environmental, psychosocial, behavioral, and developmental
influences may significantly affect long-term outcome.
A. Findings associated with increased risk of neurologic sequelae
1. Apgar score of 0-3 at 20 min of age.
2. Presence of multiorgan failure, particularly oliguria persisting beyond 24 h of life.
3. Severity of the neonatal neurologic syndrome. Severe HIE (Sarnat stage 3) carries a
mortality rate of ~80%, and survivors often have multiple disabilities, including spastic CP, severe or
profound mental retardation, cortical blindness, or seizure disorder (Robertson & Finer, 1993). There
is no permanent sequelae for mild HIE (Sarnat stage 1). Moderately affected (stage 2) patients have
outcomes that vary with their overall clinical course and duration of their neurologic condition. Stage
2 beyond 5 days is a poorer prognostic sign.
4. Duration of neonatal neurologic abnormalities. Disappearance of neurologic abnormalities
by 1-2 weeks and the ability to nipple feed normally is an excellent prognostic sign.
5. Presence of neonatal seizures, especially if they occur within the first 12 h after birth and are
difficult to control.
6. An abnormal MRI obtained in the first 24-72 h is associated with a poor outcome,
irrespective of birth variables. On the other hand, a normal MRI obtained in the first 24-72 h almost
always predicts a favorable outcome, even in a severely asphyxiated infant (Martin & Barkovich,
1995). An abnormal signal in the posterior limb of the internal capsule predicted an unfavorable
outcome in 33 of 36 infants with Sarnat stage 2 HIE (Rutherford et al, 1998). The prognostic value is
improved by repeating the study after several months, when delayed myelinization and structural
damage are better appreciated.
7. Severity and duration of EEG abnormalities. Normal to mildly abnormal EEG patterns
within the first days after delivery are significantly correlated with normal outcomes, and moderately
to severely abnormal EEG patterns are significantly related to abnormal outcomes (van Lieshout et al,
1995). A burst-suppression or isoelectric pattern on any day and prolonged EEG depression after day
12 are associated with a poor outcome. Recovery of normal EEG background by day 7 is associated
with a normal outcome. The early presence (within the first days after birth) of a normal or near-
normal EEG, even in a "comatose" child, is a strong predictor of a good neurologic outcome.
8. Persistent abnormalities of brainstem function are generally incompatible with long-term
survival.
9. Abnormal visual, auditory, or somatosensory evoked potentials persisting beyond day 7 of
life. Normal somatosensory evoked potentials (SSEPs) are highly predictive of a normal outcome.
Eken et al (1995) showed that SSEPs performed within 6 h after delivery had a positive predictive
value of 82% for moderate to severe HIE and a negative predictive value of 92%. Abnormal visual
evoked potential (VEP) throughout the first week of life or an absent VEP at anytime guaranteed an
abnormal outcome in asphyxiated full-term infants (Muttitt et al, 1991).
10. Subsequent hearing is normal in most children who have suffered perinatal or
postnatal asphyxia. Children with residual neurodevelopmental deficits have more frequent
peripheral hearing loss and more abnormalities of the central components of auditory evoked
potentials than those who do not have neurodevelopmental deficits, suggestive of residual
dysfunction in the rostral brainstem (Jiang, 1995; Jiang & Tierney, 1996).
11. Microcephaly at 3 months of age is predictive of poor neurodevelopmental outcome
(Shankaran et al, 1991). A decrease in head circumference (HC) ratios (actual HC/mean HC for age ×
100%) of >3.1% between birth and 4 months of age is highly predictive of the eventual development
of microcephaly before 18 months of age (Cordes et al, 1994). Suboptimal rate of head growth
associated with moderate cerebral white matter changes on MRI may be a better predictor of poor
neurodevelopmental outcome (Mercuri et al, 2000).
12. Decreased cerebral concentrations of phosphocreatine or ATP at birth on quantitative
31
P MRI (Martin et al, 1996).
13. Elevated brain lactate levels (Leth et al, 1996), elevated ratio of lactate to N-
acetylaspartate(Penrice et al, 1996) and lactate to choline (Barkovich et al, 1999) on proton MRS,
and low CSF cyclic adenosine monophosphate (cAMP) levels (Pourcyrous et al, 1999).
14. Increased CBF on Doppler sonography in the first 3 days after birth (Leth et al, 1996).
15. Decreased cerebral resistive index on Doppler sonography (Gonzalez de Dios et al, 1995).
16. The presence of optic atrophy is an indicator of poor visual outcome (Luna et al, 1995).
Many children with postasphyxial CNS abnormalities have lower visual acuity scores and smaller
visual fields.
B. Nondisabled survivors of moderate HIE have delayed skills in reading, spelling, or arithmetic
and have more difficulties with attention and short-term recall than survivors of mild HIE and normal
individuals.
X. Ethics. Decision making is often difficult, but it is easier if the medical team and families