Thể loại bất thường bẩm sinhva di truyen
Bạn đang xem bản rút gọn của tài liệu. Xem và tải ngay bản đầy đủ của tài liệu tại đây (513.31 KB, 32 trang )
ể loại:Bất thường bẩm sinhva di truyen
Mục lục
1
2
3
4
5
6
Bất thường bẩm sinh
1
1.1
1
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Bại não
2
2.1
Lịch sử . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2
2.2
Nguyên nhân bại não . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2
2.3
Tần suất . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3
2.4
Các thể bại não . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3
2.5
Chẩn đoán . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4
2.6
Điều trị
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4
2.7
Phòng ngừa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
5
2.8
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
5
Hiện tượng thoái hóa cột sống bẩm sinh
6
3.1
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
6
3.2
Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
6
Hội ứng người cá
7
4.1
7
Chú thích . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Hội ứng tetra-amelia
8
5.1
Đặc điểm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8
5.2
Nguyên nhân và di truyền . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8
5.2.1
Gen WNT3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8
5.2.2
Di truyền học trong gia đình . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9
5.3
Dịch tễ học . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9
5.4
Một số người mắc hội chứng tetra-amelia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9
5.5
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10
5.6
Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10
Hội ứng VACTERL
11
6.1
Từ ngữ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
11
6.2
Xem thêm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
11
6.3
am khảo và liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12
i
ii
MỤC LỤC
6.4
7
8
9
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12
Sứt môi và hở hàm ế
13
7.1
Dịch tễ học
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13
7.2
Phân dạng . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13
7.3
Điều trị
13
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3.1
Sơ sinh
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3.2
Phẫu thuật
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13
7.3.3
Nha khoa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
14
7.3.4
Phát âm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
14
13
7.4
Chú thích
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
14
7.5
Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
14
alidomide
15
8.1
Công dụng . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15
8.2
Tác hại đến thai . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15
8.3
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15
8.4
Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
15
Bệnh di truyền
16
9.1
16
am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10 Bệnh tan máu bẩm sinh
17
10.1 Phân loại bệnh alassemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
10.2 Triệu chứng của bệnh . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
10.3 Điều trị . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
10.4 Khả năng di truyền . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
10.5 ử nghiệm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18
10.6 Phòng ngừa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18
10.7 Những nghiên cứu đang được tiến hành . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18
10.8 am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
19
10.9 Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
19
11 Hội ứng Down
20
11.1 am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12 Hội ứng Klinefelter
20
21
12.1 Nguyên nhân . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
21
12.2 Sinh lý bệnh . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
21
12.3 Dấu hiệu và triệu chứng . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
22
12.4 Chẩn đoán trước khi sinh
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
22
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
23
12.5 Điều trị
12.6 Biến chứng
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
23
12.7 Tiên lượng . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
23
MỤC LỤC
iii
12.8 Biến thể . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
23
12.9 am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
24
12.10 Xem thêm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
24
12.11 Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
24
13 Rối loạn sắc giác
25
13.1 Chú thích . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
25
13.2 Đọc thêm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
25
13.3 Liên kết ngoài . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
25
14 Trimethylaminuria
26
14.1 am khảo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
26
14.2 Nguồn, người đóng góp, và giấy phép cho văn bản và hình ảnh . . . . . . . . . . . . . . . . . . .
27
14.2.1 Văn bản . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
27
14.2.2 Hình ảnh . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
27
14.2.3 Giấy phép nội dung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
28
Chương 1
Bất thường bẩm sinh
1.1 Tham khảo
Bất thường bẩm sinh (tiếng Anh: congenital disorder)
là tên gọi chung của các bệnh có sẵn khi sinh ra. Nhiều
bệnh có thể được chẩn đoán trước khi sinh hay vừa sinh
ra trong khi một số bệnh khác chỉ biểu hiện nhiều năm
sau khi sinh. Bệnh bất thường bẩm sinh có thể do bất
thường di truyền, tai biến trong tử cung hay rối loạn
trong quá trình hình thành của phôi, thai. Nhiều bệnh
bất thường bẩm sinh vẫn chưa biết nguyên nhân.
Bệnh bất thường bẩm sinh (BTBS) bao gồm những bất
thường từ nhẹ như vảy đỏ trên da cho đến nặng như
bệnh tim bẩm sinh hay sinh ra không có tay, chân.
Nhiều bệnh ảnh hưởng đến chuyển hóa trong cơ thể.
Những bất thường trầm trọng (như não không hình
thành) làm tử vong trước khi sinh.
Bệnh BTBS đứng đầu các bệnh gây tử vong trẻ sơ sinh
(2/1000 - theo thống kê tại Hoa Kỳ).
Khoảng 2 - 3% trẻ sơ sinh bị BTBS đáng kể. Bất thường
nhiều nhất ở não (1%), tim(0,8%), thận (0,4%) và tứ chi
(0,2%). Trong những bệnh bẩm sinh gây tử vong cho trẻ
sơ sinh, bệnh tim đứng đầu (28%), theo sau là bệnh bất
thường nhiễm sắc thể và đường phổi (15%) và bệnh não
(12%).
Khoảng 25% BTBS là do bất thường của hệ thông tin
di truyền - nghĩa là thay đổi bất thường trong gene.
Chừng 5% bị bất thường trong nhiều hoặc hết thảy
nhiễm sắc thể (chromosome). Nhiều BTBS xảy ra vì cha
hay mẹ có mang mầm bệnh trong gene, một số khác là
do biến dạng của chromosome trong thời phôi thai.
Ảnh hưởng từ môi trường bên ngoài tác động vào người
mẹ cũng có thể gây ra BTBS:
• Dinh dưỡng: iếu chất folic acid (hở cột sống)
• Độc tố: ủy ngân trong thức ăn như cá biển lớn
• Dược phẩm: Phenytoin (sứt môi, hư tim);
alidomide (cụt tay)
• Nhiễm trùng hay siêu vi trùng: giang mai và sởi
Đức (mù, dị tật tim, dị tật hệ thần kinh)
Một số lớn trẻ em sinh ra bị BTBS nhưng không rõ lý
do.
1
Chương 2
Bại não
Bại não là thuật ngữ chỉ một nhóm tình trạng bệnh lý
mãn tính ảnh hưởng đến sự kiểm soát các vận động
cũng như tư thế. Do một phần nào đó của bộ não bị tổn
thương nên trẻ bệnh không thể cử động các cơ được
vùng não đó điều khiển một cách bình thường được.
Các triệu chứng của bại não có thể nhẹ nhàng hoặc
rất nặng nề ở các trẻ khác nhau tùy theo tổn thương
não nhưng ở một trẻ nhất định thì triệu chứng không
nặng lên khi trẻ lớn hơn. Nói một cách khác, bại não
là một bệnh tĩnh, nghĩa là các tổn thương đã định hình
và không tiến triển xấu hơn nữa. Định nghĩa này rất
quan trọng để phân biệt bại não với các tình trạng tổn
thương thần kinh khác có tổn thương não hoạt động
và do đó triệu chứng tâm thần vận động sẽ càng ngày
càng nặng hơn. Nếu được điều trị, phần lớn trẻ bị bại
não có những biến chuyến rất khả quan.
này có thể bắt nguồn từ rất sớm trong quá trình phát
triển của não bộ khi trẻ còn đang trong giai đoạn bào
thai.
Mặc dù có những quan sát và nhận định tinh tế của
Freund, cho mãi đến gần đây rất nhiều thầy thuốc, gia
đình thậm chí cả các nhà nghiên cứu vẫn tin tưởng là
thiếu ôxy não là nguyên nhân bại não. Tuy nhiên vào
thập niên 1980 nhờ vào những nghiên cứu quy mô lớn
và với những phương pháp mới, các nhà khoa học nhận
định chắc chắn rằng biến chứng của sinh khó chỉ chiếm
10% tổng số các trường hợp bại não. Trong phần lớn
các trường hợp bại não, người ta vẫn chưa thể xác định
được nguyên nhân. Các nghiên cứu vẫn đang được tiến
hành.
Rất nhiều trẻ bị bại não thường có kèm theo các tình 2.2 Nguyên nhân bại não
trạng bệnh khác đòi hỏi phải điều trị. Các bệnh này
gồm chậm phát triển tâm thần, rối loạn khả năng học Trong khoảng 70% trường hợp, bại não có thể là do
tập, động kinh, các vấn đề về thính giác, thị giác, ngôn những bất thường xảy ra trước sinh làm ảnh hưởng đến
ngữ.
quá trình phát triển bình thường của não. eo báo cáo
năm 2003 của Hội Sản và Phụ khoa Hoa Kỳ (American
College of Obstetricians and Gynecologists - ACOG) và
Viện Hàn lâm Nhi khoa Hoa Kỳ (American Academy
2.1 Lịch sử
of Pediatrics – AAP) thì thiếu ôxy trong quá trình sinh
đẻ chỉ chiếm một tỷ lệ nhỏ trong các trường hợp bại
Vào năm 1860, William Lile, một phẫu thuật viên
não. Mặc dù nhiều trường hợp người ta chưa thể xác
chỉnh hình người Anh đã cho xuất bản những bài báo
định được căn nguyên nhưng các nguyên nhân được
đầu tiên về một rối loạn khó hiểu ảnh hưởng đến trẻ
biết của bại não bao gồm:
em trong những năm đầu đời gây nên co cứng rõ các
cơ ở chân và ở tay nhưng mức độ nhẹ hơn. Những đứa
• Nhiễm trùng trong thai kỳ
trẻ này có khó khăn trong cầm nắm đồ vật, bò và đi
lại. Những rối loạn này không cải thiện khi trẻ lớn lên
nhưng cũng không nặng nề hơn. Tình trạng này đầu Các nhiễm trùng ở phụ nữ có thai như rubella (sởi
tiên được gọi là bệnh Lile trong nhiều năm. Những Đức), cytomegalovirus và toxoplasmosis có thể gây tổn
đứa trẻ này dường như sinh non hoặc do biến chứng thương não của bào thai và gây bại não sau này. Các
trong quá trình sinh nở nên Lile đưa ra giả thiết là nhiễm trùng khác như nhiễm trùng ối, nhiễm trùng hệ
chứng bệnh này là hậu quả của tình trạng thiếu ôxy não tiết niệu – sinh dục của người mẹ cũng có thể gây nên
trong lúc sinh. Ông ta cho rằng sự thiếu ôxy này đã làm sinh non, một nguy cơ khác của bại não.
tổn thương những vùng não nhạy cảm có chức năng
• iếu khí não bào thai
kiểm soát vận động. Tuy nhiên vào năm 1897, nhà tâm
lý học lừng danh người Áo Sigmund Freund đã không
tán thành giả thiết này. Do quan sát thấy những trẻ này Khi chức năng của nhau thai bị giảm sút (suy nhau thai)
có các rối loạn khác như chậm phát triển tinh thần, rối hoặc bị bóc tách khỏi thành tử cung trước khi sinh
loạn thị lực và động kinh nên Freund cho rằng rối loạn (nhau bong non) hoặc do chảy máu do sai lệch vị trí
2
2.3. TẦN SUẤT
(nhau tiền đạo) có thể làm giảm lượng ôxy cung cấp
cho thai nhi.
• Sinh non
Sinh non là trẻ sinh ra trước 37 tuần thai tính từ ngày
đầu tiên của kỳ kinh nguyệt cuối cùng trước khi có
thai. Những trẻ sinh non đặc biệt trước 32 tuần và nhất
là trước 28 tuần thai có nguy cơ bại não rất cao. Một
nghiên cứu cho thấy những trẻ sinh non có cân nặng
lúc sinh thấp hơn 1500 gram có nguy cơ bại não cao
gấp 30 lần so với trẻ sinh đủ tháng (trẻ sinh từ 37 đến
42 tuần thai). Lý do là trẻ sinh non có nguy cơ rất cao bị
xuất huyết não gây tổn thương các tổ chức mong manh
đang phát triển của não hoặc gây nên chứng nhuyễn
hóa chất trắng quanh não thất.
• Ngạt trong quá trình uyển dạ và sinh nở
3
• Bại não mắc phải
Trẻ mắc các chứng bệnh gây tổn thương thần kinh
trong hai năm đầu tiên của đời sống ví dụ như viêm
màng não mủ, viêm não, chấn thương sọ não…
2.3 Tần suất
Bại não thường ít khi được chẩn đoán sớm trước 2 tuổi.
Với lứa tuổi trên 3 thì tần suất bại não vào khoảng 2-3
trường hợp/1000 trẻ. Đây là một tỷ lệ khá cao đối với
một bệnh mãn tính như thế này. Ngay cả ở Mỹ thì cũng
có khoảng trên nửa triệu bệnh nhân bại não.
2.4 Các thể bại não
Cho mãi đến gần đây người ta vẫn còn tin tưởng rộng Bại não được chia thành 3 thể lâm sàng chính. Tuy
rãi là ngạt (thiếu ôxy) trong quá trình chuyển dạ và sinh nhiên trên một trẻ bại não có thể có nhiều hơn một
nở là nguyên nhân của hầu hết các trường hợp bại não. thể bệnh kết hợp với nhau.
Tuy nhiên như trên đã nói, theo nghiên cứu của Hội
Sản và Phụ khoa Hoa Kỳ và Viện Hàn lâm Nhi khoa
• Bại não thể liệt cứng (spastic cerebral palsy): Có
Hoa Kỳ thì ngạt chỉ chiếm 10% trong tổng số các bệnh
khoảng 70 đến 80% bệnh nhân bại não thuộc nhóm
nhân bại não.
này. Trẻ mắc thể này có biểu hiện các cơ co
cứng,luôn ở trạng thái tăng trương lực cơ. Chính
do tình trạng này mà sự vận động của bệnh nhân
• Các bệnh máu
bại não rất khó khăn. Trẻ khó cầm nắm, bò hoặc
đi. ể lâm sàng này lại được chia làm ba nhóm
Bất đồng nhóm máu Rh là sự bất tương hợp nhóm
nhỏ. Trong nhóm liệt cứng hai i dưới (spastic
máu giữa mẹ và bào thai gây nên vàng da trầm trọng
diplegia), trẻ có bất thường co cứng rõ ở hai chi
và tổn thương não dẫn đến bại não. Bệnh này thường
dưới. Do các cơ khép co cứng nên chân trẻ luôn bị
gặp ở người da trắng còn ở Việt Nam rất hiếm gặp vì
kéo vào trong làm cho trẻ có dáng đi bắt chéo hai
tỷ lệ mang Rh (-) cực kỳ hiếm gặp. Tuy nhiên ở Việt
chân rất đặc trưng. Nhóm thứ hai gồm những trẻ
Nam có thể gặp bất đồng nhóm máu ABO giữa mẹ và
bị liệt cứng nửa người (spastic hemiplegia) thường
thai nhi. Một bệnh khác rất nặng nề mặc dù biện pháp
có biểu hiện liệt cứng một bên (phải hoặc trái).
phòng ngừa cực kỳ đơn giản là xuất huyết não do thiếu
ường thì chi trên bị ảnh hưởng nặng hơn chi
Vitamin K ở trẻ sơ sinh và nhũ nhi cũng gây nên bại
dưới. Nặng nề nhất là nhóm trẻ liệt cứng tứ i
não. Các bệnh rối loạn chức năng đông máu khác cũng
(spastic quadriplegia). Bệnh nhân thuộc nhóm này
có thể là nguyên nhân của bại não vì làm tăng nguy cơ
có biểu hiện liệt cứng cả hai chi trên và hai chi dưới
chảy máu trong não.
cùng với các cơ trục thân. Cả các cơ ở mặt cũng bị
ảnh hưởng làm cho trẻ bị tàn phế rất nặng.
• Vàng da nhân
Vàng da trẻ sơ sinh là do sự tích tụ trong máu một loại
sắc tố có tên billirubin do tốc độ phá hủy hồng cầu cao
và chức năng gan chưa trưởng thành ở trẻ sơ sinh, đặc
biệt là trẻ đẻ non. Trong trường hợp nặng, sắc tố này có
thể vượt qua hàng rào mạch máu – não và lắng đọng
chủ yếu ở các nhân nền của não (do đó có tên là vàng
da nhân) và làm tổn thương các cấu trúc này đưa đến
thể bại não kèm múa vờn.
• Các bất thường bẩm sinh khác
Các trẻ có bất thường cấu trúc hệ thần kinh, nhiều bệnh
di truyền khác cũng làm tăng nguy cơ bại não.
• Bại não thể múa vờn hay loạn động (athetoid hay
dyskinetic cerebral palsy): Có khoảng 10 đến 20%
bệnh nhân bại não thuộc vào nhóm này. Đây là
thể bệnh được đặc trưng bằng sự thay đổi thất
thường của trương lực cơ (lúc tăng, lúc giảm). Trẻ
còn thường kèm theo các động tác bất thường
không kiểm soát được. Các động tác này có nhịp
điệu chậm, biên độ đôi khi rộng nhưng đang múa
nhưng trẻ không ý thức được điều này. Do bất
thường trong kiểm soát cử động như vậy nên bệnh
nhân khó có tư thế ngồi hoặc dáng đi bình thường.
Ngoài ra các cơ ở mặt và lưỡi cũng bị ảnh hưởng
nên trẻ khó bú (với trẻ còn bú) hoặc khó nuốt, khó
nói.
4
CHƯƠNG 2. BẠI NÃO
• Bại não thể thất điều (ataxic cerebral palsy):
Khoảng 5 đến 10% bệnh nhân bại não thuộc thể
lâm sàng này. Bệnh ảnh hưởng chủ yếu đến cân
bằng tư thế và phối hợp động tác. Do có rối loạn
trong kiểm soát tư thế nên dáng đi của trẻ hay lảo
đảo, vùng thắt lưng hay đong đưa. Do rối loạn khả
năng phối hợp động tác nên trẻ rất khó thực hiện
được các động tác đòi hỏi sự nhịp nhàng như vỗ
tay theo nhịp hoặc đòi hỏi độ chính xác như viết.
2.5 Chẩn đoán
• Bối cảnh phát hiện
Nếu trong thai kỳ, đặc biệt vào những tháng đầu tiên,
mẹ bị các bệnh như cúm, sởi Đức, hoặc dùng một số
thuốc có khả năng gây quái thai hoặc ảnh hưởng đến sự
phát triển bình thường của ống thần kinh thì cần theo
dõi đặc biệt. Những trẻ có tiền sử sinh non, ngạt chu
sinh cũng là những đối tượng có nguy cơ cao. Tuy nhiên
các bậc cha mẹ có thể theo dõi sự phát triển của con
mình như lật, ngồi, bò, đi nếu có nghi ngờ bất thường
thì cần có ý kiến của bác sĩ chuyên khoa nhi ngay.
• Khám lâm sàng
Mặc dù nguyên nhân bại não là do những sự kiện xảy ra
trong thai kỳ và trong hai năm đầu đời sống, việc đánh
giá và chẩn đoán bại não trước hai tuổi rất khó khăn.
Khám trương lực cơ, cơ lực đòi hỏi phải có chuyên môn
sâu về thần kinh nhi khoa. Trương lực cơ có thể tăng
hoặc giảm. Các phản xạ nguyên thủy thường mất đi
sau 6 tháng nhưng ở trẻ bại não thì các phản xạ này
tồn tại lâu hơn. Trẻ nhỏ trước 12 tháng thường không
biểu hiện rõ thuận tay nào. Nhưng đối với trẻ bị bại
não (nhất là thể liệt cứng nửa người) thì khuynh hướng
thuận tay xuất hiện sớm (do bên liệt vận động khó,
trẻ phải vận động bên lành). Đánh giá sự phát triển
tâm thần vận động thường có thể dựa vào các tiêu
chuẩn như tiêu chuẩn Amiel-Tison hoặc thang đánh
giá Denver.
• Xét nghiệm hỗ trợ
Các xét nghiệm chẩn đoán hình ảnh như siêu âm
não qua thóp, chụp cắt lớp vi tính (CT: Computerised
Tomography) đặc biệt là chụp cộng hưởng từ (MRI:
Magnetic Resonnance Imaging) cho biết những thông
tin giá trị về tổn thương não. Các xét nghiệm hóa sinh
hay di truyền tùy theo hướng chẩn đoán trên lâm sàng.
Đo điện não đồ (EEG: ElectroEncephaloGram) cũng là
một xét nghiệm cơ bản không thể thiếu trong chẩn
đoán bại não cũng như các bệnh của hệ thần kinh trung
ương khác.
2.6 Điều trị
Điều trị bại não cần có sự phối hợp của nhiều chuyên
ngành khác nhau cùng phối hợp với trẻ và gia đình
nhằm vạch ra được một kế hoạch cụ thể thích hợp cho
từng cá nhân. Điều trị bại não nhằm giúp trẻ đạt được
khả năng trí tuệ cũng như vận động tối đa có thể có
chứ không thể lấy lại được những khả năng đã mất để
thành một đứa trẻ hoàn toàn bình thường. Các chuyên
khoa liên quan bao gồm nhi khoa, phục hồi chức năng,
phẫu thuật chỉnh hình, mắt, tâm thần, chỉnh âm, dạy
nghề… Nếu điều trị được thực hiện sớm và có sự phối
hợp chặt chẽ của các chuyên ngành, đặc biệt là sự tham
gia của gia đình thì kết quả rất khả quan.
• Trẻ thường được bắt đầu bằng vật lý trị liệu ngay
sau khi được chẩn đoán. Điều trị này làm tăng kỹ
năng vận động của trẻ như ngồi, đi, cải thiện cơ
lực và phòng ngừa sư co kéo biến dạng cơ (cơ bị
co rút có thể giới hạn vận động của các khớp). Đôi
khi trẻ còn được sử dụng các dụng cụ như nẹp,
máng hoặc bó bột để ngừa co rút cơ và cải thiện
chức năng của chân và tay. Nếu tình trạng co rút
cơ quá nặng, trẻ cần được phẫu thuật chỉnh hình
để làm dài cơ bị bệnh.
• Đôi khi thầy thuốc còn dùng một số thuốc nhằm
làm giảm bớt mức độ co cứng của cơ và làm giảm
các cử động bất thường. Tuy nhiên các thuốc
đường uống hiện nay không có tác dụng đáng kể.
Có thể tiêm trực tiếp Botox (botulinum toxin) vào
các cơ co rút có thể giúp cải thiện triệu chứng, tác
dụng có thể kéo dài vài tháng (trong thời gian này
việc phục hồi chức năng thực hiện dễ dàng hơn).
Một loại thuốc khác cũng chứng tỏ tác dụng tốt
đối với các trường hợp liệt cứng mức độ vừa đến
nặng. uốc này có tác dụng chống co cơ có tên
là baclofen. Để đưa thuốc vào bệnh nhân, người ta
phải phẫu thuật để đưa vào dưới da một bơm tiêm,
thông qua đó thuốc được bơm liên tục.
• Đối với một số trẻ có tình trạng co cứng hai chi
dưới nặng nề, phẫu thuật cắt bỏ chọn lọc một số
nhánh thần kinh ở lưng chi phối hoạt động chi có
thể làm giảm vĩnh viễn tình trạng co cứng cũng
như cái thiện khả năng vận động như ngồi, đứng,
đi. Phẫu thuật này thường tiến hành khi trẻ được
2 đến 7 tuổi.
• Đối với trường hợp nặng, tập luyện cho trẻ nhằm
mục đích thực hiện được các thao tác quan trọng
nhất trong cuộc sống hằng ngày như ăn, mặc,
vệ sinh cá nhân. Những trường hợp nhẹ hơn, tập
luyện và điưều trị có thể hướng đến mục tiêu cao
hơn như giao tiếp, vui chơi, và cả học tập nữa.
• Điều quan trọng quyết định sự thành công của
điều trị là vai trò của bố mẹ hoặc người chăm sóc
trẻ. Nếu những đối tượng này tin tưởng, quyết tâm
2.8. THAM KHẢO
và có những kiến thức cơ bản cũng như kỹ năng
thực hành thì đứa trẻ có nhiều khả năng đạt được
những tiến bộ lớn nhằm giảm sự lệ thuộc của trẻ
vào người khác, đảm bảo cho trẻ một cuộc sống
gần với bình thường.
2.7 Phòng ngừa
Hiện nay y học vẫn chưa hiểu hết các nguyên nhân gây
bại não do đó việc phòng ngừa không đạt hiệu quả như
mong muốn.
• Nâng cao chất lượng chăm sóc phụ nữ trong tuổi
sinh đẻ và phụ nữ có thai nhằm giảm thiểu những
biến chứng của thai kỳ. Phân tuyến để điều trị
sản khoa hợp lý nhằm giảm thiểu các biến chứng
do sinh đẻ như ngạt, chấn thương… Có trung tâm
chăm sóc sơ sinh phù hợp.
• Tiêm ngừa đề phòng các bệnh như viêm màng não
mủ, viêm não
• Phòng chống tai nạn giao thông cũng như các tai
nạn khác (ngạt nước…)
• Phòng ngừa thứ phát là phát hiện sớm và điều trị
trẻ bị bại não nhằm hạn chế tật nguyền.
• Cerebral Palsy
2.8 Tham khảo
5
Chương 3
Hiện tượng thoái hóa cột sống bẩm sinh
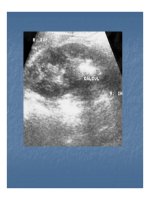
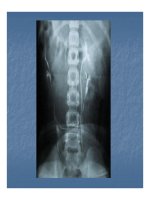
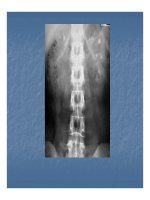
Có thể chẩn đoán bệnh từ phương pháp Chụp cộng
hưởng từ và chụp cắt lớp quang tuyến (X-ray computed
tomography).
3.1 Tham khảo
3.2 Liên kết ngoài
• (tiếng Anh)
• (tiếng Anh) Caudal Regression Syndrome and
Sirenomelia in Only One Twin in Two Diabetic
Pregnancies
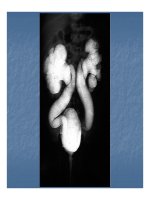
Hội ứng thoái hóa phần dưới (cột sống) hay Hiện
tượng thoái hóa cột sống bẩm sinh (tiếng Anh: Caudal
regression syndrome) là một hội chứng rối loạn hay bất
thường bẩm sinh hiếm thấy khi có sự phát triển bất
thường của cột sống (xương sống) của thai nhi.
Rối loạn này có thể dẫn đến một loạt các vấn đề y học
khác nhau, từ thiếu một phần của vùng xương đuôi của
cột sống cho đến nhiều trường hợp nghiêm trọng hơn
liên quan đến biến đổi dị dạng của xương chậu và phần
dưới cột sống, tủy sống. Một số trường hợp nhỏ mà
không gây ra triệu chứng gì, trong khi các trường hợp
nghiêm trọng hơn có thể đi kèm với khuyết tật (thiếu
chân) hay tê liệt chân và bất thường bẩm sinh lớn, suy
thần kinh và không có khả năng điều hóa hệ bài tiết
hay tiểu tiện.
6
Chương 4
Hội chứng người cá
chứng thoái hóa phần dưới) và hội chứng người cá,[3]
tuy nhiên sự liên quan này nói chung không được chấp
nhận.[4]
VACTERL-H là sự mở rộng của Hội chứng VACTERL
bao gồm chẩn đoán về hình thức ít nghiêm trọng của
hội chứng người cá.[5] Trước đây, rối loạn này được xếp
vào loại Caudal regression syndrome; tuy nhiên, nó đã
phân loại lại để tách ra thành trường hợp khác.
4.1 Chú thích
[1] Kallen B, Castilla EE, Lancaster PA, Mutchinick O,
Knudsen LB, Martinez-Frias ML, Mastroiacovo P,
Robert E (1992). “e cyclops and the mermaid:
an epidemiological study of two types of rare
malformation”. J Med Genet 29 (1): 30–5. PMID
1552541. doi:10.1136/jmg.29.1.30.
[2] Mary Beth Sammons. “"Shiloh Pepin: e Mermaid
Girl"”.
[3] Abraham M. Rudolph, Robert K. Kamei and Kim
J. Overby. “"Rudolph’s Fundamentals of Pediatrics"”.
McGraw Hill Professional 2002, ISBN 0838584500. Truy
cập ngày 21 tháng 4 năm 2008.
Sirene (Sirenomelia), Viện bảo tàng lịch sử và xác ướp, Pháp.
[4] Oneije C. I., Sherer D. M., Handwerker S. and Shah L.
“"Caudal Regression Syndrome and Sirenomelia in Only
One Twin in Two Diabetic Pregnancies"”. Clinical and
Experimental Obstetrics and Gynecology 31: 151-3; 2004.
Truy cập ngày 17 tháng 4 năm 2008.
Hội ứng người cá hay còn gọi là Sirenomelia, nó là
những trường hợp hiếm của bất thường bẩm sinh có
hai chân bị dính lại với nhau giống như cái đuôi của cá
và làm họ giống như người cá, vì lý do đó tên của hội
chứng này là hội chứng người cá.
[5] E Assimakopoulos, A Athanasiadis, M Zafrakas, K
Dragoumis and J Bontis. “"Prenatal diagnosis of
sirenomelia with bilateral hydrocephalus: Report of
a previously undocumented form of VACTERL-H
association"”. American journal of perinatology 15: 19397; 1998. Truy cập ngày 21 tháng 4 năm 2008.
Trường hợp này xảy ra với tỉ lệ 1/100.000[1] (hiếm giống
như trường hợp Sinh đôi dính liền) và thường thường
thì mạng sống chỉ kéo dài từ một đến hai ngày do biến
chứng liên quan đến sự phát triển và chức năng của
thận và bọng đái. Hơn một nửa trường hợp “hội chứng
người cá" dẫn đến thai chết non và tình trạng này có
khả năng xảy ra gấp hơn 100 lần trong các ca sinh đôi
giống hệt nhau (sinh đôi cùng trứng) so với sinh đơn
hay sinh đôi khác trứng.[2] Kết quả này là từ sự hư hại
của mạch máu cung cấp từ động mạch chủ phía dưới
trong tử cung. Bệnh tiểu đường của người mẹ được
cho là có liên quan đến caudal regression syndrome (Hội
7
Chương 5
Hội chứng tetra-amelia
Hội ứng tetra-amelia (tetra- + amelia), còn được gọi
là nhiễm sắc thể lặn tetraamelia hay hội ứng thiếu
tứ i,[1] là sự di truyền nhiễm sắc thể lặn rất hiếm [2]
đặc trưng bởi bất thường bẩm sinh với sự thiếu hụt các
chi (4 chi trên cơ thể).[3] Các bộ phận khác của cơ thể
cũng bị ảnh hưởng bởi dị tật như mặt, sọ, cơ quan sinh
dục, hậu môn và xương chậu.[1] Nguyên nhân của rối
loạn này là do đột biến gen WNT3.[2][4]
5.1 Đặc điểm
Bệnh nhân mắc phải hội chứng này bị mất (hoặc không
đầy đủ) chân tay. Ngoài ra, hội chứng tetra-amelia còn
gây ra dị tật nghiêm trọng trên các bộ phận khác nhau
của cơ thể, bao gồm mặt (sứt môi), mắt (cườm thủy tinh
thể mắt), đầu, tim, hệ thần kinh trung ương, xương, hậu
môn và cơ quan sinh dục.[5]
Trong nhiều trường hợp, phổi kém hoặc không phát
triển, do đó làm cho bệnh nhân thở rất khó khăn hoặc
không thể hô hấp. Trẻ có hội chứng tetra-amelia gặp
phải vấn đề y tế rất nghiêm trọng, vì vậy đa số chết
non hoặc chết ngay sau khi sinh. Việc quản lý những
người sống sót (chưa được thống kê) phụ thuộc sự hiện
diện và mức độ nghiêm trọng của dị tật cần có sự chăm
sóc y tế.[5] Diễn giả Nick Vujicic [6] là một trong những
nạn nhân của hội chứng sống sót sau khi sinh và nhà
văn, nhà báo thể thao Nhật Bản Hirotada Ototake là
một trường hợp khác.[7]
5.2 Nguyên nhân và di truyền
5.2.1
Mô hình hội chứng Terea-amelia di truyền với một gen tính trạng
lặn.
Gen WNT3
Các nhà nghiên cứu đã tìm thấy một đột biến ở gen
WNT3 ở người với hội chứng tetra-amelia từ một đại
gia đình.[8] Gen này là một phần của họ gen WNT,
đóng vai trò chính trong sự phát triển sau khi sinh. Gen
WNT3 nằm ở nhiễm sắc thể 17q21.[9]
ngăn cản các tế bào sản xuất chức năng WNT3 protein,
Các protein sinh ra từ gen WNT3 có liên quan đến sự phá vỡ hình thành chân tay bình thường và dẫn đến
hình thành của các chi và các cơ quan cơ thể khác trong dị tật bẩm sinh nghiêm trọng khác liên quan đến hội
quá trình phát triển phôi thai.[10] Đột biến ở gen WNT3 chứng tetra-amelia.
8
5.4. MỘT SỐ NGƯỜI MẮC HỘI CHỨNG TETRA-AMELIA
9
Nick Vujicic, một trong những người mắc hội chứng tetra-amelia
Nghệ sĩ Violetta, hình chụp năm 1925
5.2.2
Di truyền học trong gia đình
Trong các gia đình bị nhiễm, nguyên nhân của hội
chứng tetra-amelia chưa được xác định. Một số nhà
nghiên cứu tin rằng đột biến chưa nhận dạng được ở
gen WNT3 hay các gen khác có liên quan đến sự phát
triển các chi cơ thể có thể chịu trách nhiệm cho rối loạn.
Ở vài gia đình được báo báo, hội chứng tetra-amelia
xuất hiện với một mẫu di truyền tính trạng lặn.[1][2]
Điều này có nghĩa gen khiếm khuyến chịu trách nhiệm
cho sự rối loạn nằm ở nhiễm sắc thể thứ 17, và hai bản
sao của gen khiếm khuyết gen (từ cha và mẹ) tạo thành
cặp gen rối loạn khi sinh. Cha mẹ chỉ mang một gen
khiếm khuyết thông thường sẽ không bị rối loạn hay
ảnh hưởng bởi hội chứng này. Nếu cha và mẹ mang 1
gen bị nhiễm, con cái sinh ra sẽ có 25% cơ hội không bị
nhiễm, 50% là cá thể mang gen bị nhiễm và 25% là cá
thể mắc hội chứng này.[5]
số đó là một người ở Philippines và một ở Anh. Một
người khác cũng mắc hội chứng này và không có chân
tay là Joanne O'Riordan ở Millstreet, Cork, Ireland. Cô
đã được ủ tướng Ireland Enda Kenny tiếp đón, nói
chuyện tại Liên Hiệp ốc (ở tuổi 16), và xuất hiện
trước hội nghị “iếu nữ trong công nghệ" của Liên
minh Viễn thông ốc tế, và được hoan nghênh nhiệt
liệt sau khi có bài phát biểu. Cô cũng thảo luận với
trường Học viện Công nghệ Massachuses và công ty
Apple Inc.[12]
5.4 Một số người mắc hội chứng
tetra-amelia
• Marilee Adamski-Smith [13]
• Christian Arndt [14]
5.3 Dịch tễ học
Hội chứng tetra-amelia là một rối loạn rất hiếm gặp và
cho đến nay chỉ được miêu tả ở năm gia đình thuộc các
chủng tộc khác nhau (Ả Rập, Maroc, Syria-Aramaic).
Hiện không có báo cáo ước tính về tỷ lệ và tần số dân
số mang hội chứng tetra-amelia.[5] Chỉ có vài người
trên thế giới được biết là mắc hội chứng này,[11] trong
• Joanne O'Riordan [15]
• Hirotada Ototake [16]
• Prince Randian
• Yovana Yumbo Ruiz [17]
• Violea (entertainer) [18]
10
CHƯƠNG 5. HỘI CHỨNG TETRA-AMELIA
• Nick Vujicic
• Piotr Radoń
5.6 Liên kết ngoài
[19]
• Roberts Syndrome, NCBI.
5.5 Tham khảo
[1] 'Dự án di truyền Mendel ở người' (trực tuyến) (OMIM)
273395
[2] PMID 14872406 (PMID 14872406)
Citation will be completed automatically in a few
minutes. Jump the queue or expand by hand
[3] Tetra-amelia syndrome, Genetics Home Reference, U.S.
National Library of Medicine.
[4] Hội chứng thiếu tứ chi, Công An Nhân dân.
[5] Tetra-Amelia Syndrome, National Center for
Biotechnology Information, U.S. National Library
of Medicine.
[6] Nick Vujicic và hành trình chiến thắng số phận,
VnExpress.
[7] Limbless but limitless Ototake, ChinaPost, ngày 26
tháng 04 năm 2013.
[8] Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U,
Niswander L, Weber JL, Muller U. Homozygous WNT3
mutation causes tetra-amelia in a large consanguineous
family. Am J Hum Genet. 2004;74:558–63. PMC free
article PubMed
[9] 'Dự án di truyền Mendel ở người' (trực tuyến) (OMIM)
165330
[10] WNT3, Genetics Home Reference], U.S. National
Library of Medicine.
[11] Stephan Niemann, MD (28/8/2007). “Tetra-Amelia
Syndrome(Synonym:
Tetra-Amelia,
Autosomal
Recessive)”. Kiểm tra giá trị ngày tháng trong: |date=
(trợ giúp)
[12] Boland, Rosita (ngày 1 tháng 6 năm 2012). “Joanne
O'Riordan: 'People used to say: she’s the one with no
arms or legs. ey're nicer now'”. e Irish Times (Irish
Times Trust). Truy cập ngày 1 tháng 6 năm 2012.
[13] Woman
defines
LeaderTelegram.
power
of
determination,
[14] Christian Arndt, christian-arndt.de
[15] She beat the Budget: e extraordinary, awe-inspiring
and humbling story of Joanne O'Riordan, who was born
with no arms or legs… but with the heart of a lion,
DailyMail, ngày 11 tháng 12 năm 2011.
[16] No One’s Perfect, Ralphmag.org
[17] Nghị lực của cô bé không tay, không chân, Tuổi Trẻ.
[18] Wallace Stort, Amazing Account Of A Limbless Beauty
Show, London Life magazine, ngày 27 tháng 1 năm 1940
[19] Leer from Piotr Radoń, Fecwis.org
• “Tetra-amelia syndrome - Genetics Home
Reference”. U.S. National Library of Medicine.
Truy cập ngày 9 tháng 1 năm 2009.
• “Tetra-Amelia Syndrome -- GeneReviews -- NCBI
Bookshel”. University of Washington, Seale.
Truy cập ngày 9 tháng 1 năm 2009.
• “Tetra-Amelia-Syndrome (Birth defects &
disorders)”. I-Am-Pregnant. Truy cập ngày 9
tháng 1 năm 2009.
• “Tetraamelia
multiple
malformations
WrongDiagnosis.com”. WrongDiagnosis. Truy
cập ngày 9 tháng 1 năm 2009.
Chương 6
Hội chứng VACTERL
Hội ứng VATER (tiếng Anh: VATER syndrome) hoặc
là Tập hợp VACTERL (VACTERL association) là một
tổng hợp những khuyết tật hoặc bất thường bẩm sinh.
Lý do nó thường được gọi là một tập hợp hay liên kết,
chứ không phải là một hội chứng riêng lẻ là trong khi
tất cả các khuyết tật bẩm sinh được liệt kê tại đây nhưng
không đi sâu vào nguyên nhân cũng như không tìm
hiểu gen hay bộ gen nào đã gây ra những khuyết tật
bẩm sinh này.
Mỗi trẻ sơ sinh với khuyết tật này có thể vẫn khác
nguyên nhân với bất cứ đứa trẻ nào khác. Nguyên nhân
của những trường hợp dị tật trong liên kết này vẫn
đang tranh cãi. Tập hợp VACTERL có thể được liên kết
với các điều kiện khác tương tự như tại Hội chứng dị
tật cột sống (Klippel–Feil syndrome) và Hội chứng dị
tật tai mũi họng (còn gọi là “Hội chứng Goldenhar” Goldenhar syndrome, theo tên người tìm ra là Maurice
Goldenhar).
Không trường hợp cụ thể nào có vấn đề di truyền hay
nhiễm sắc thể đã được xác định hay chứng minh với
tổng hợp VACTERL. VACTERL thường được thấy ở
những thai nhi và trẻ sơ sinh có một số khiếm khuyết
hay rối loạn nhiễm sắc thể như Hội chứng rối loạn
nhiễm sắc thể 18 (còn gọi là Hội chứng Edwards hay
Trisomy 18) và thường xuyên hơn là được thấy ở trẻ sơ
sinh của các bà mẹ bị bệnh tiểu đường. Tuy nhiên, rất
có thể VACTERL gây ra bởi nhiều yếu tố khác.
Dị tật chi bẩm sinh (ở tay)
6.1 Từ ngữ
• E - Esophageal atresia - Rối loạn thực quản (ống
tiêu hóa) hay hệ thống tiêu hóa
VACTERL là sự kết hợp những chữ đầu trong tiếng Anh
diễn tả một số trường hợp bất thường bẩm sinh:
• R - Renal (Kidney) hay là radial anomalies - Dị tật
do thận
• V - Vertebral anomalies - Bất thường hay dị tật cột
sống
• L - Limb (Congenital disorder anomalies) - Dị tật
chi, khuyết chi (thiếu chân hay tay)
• A - Anal atresia - Dị tật ở mông hay hậu môn
• C - Cardiovascular anomalies - Dị tật do tim mạch,
thường là do 1 lỗ thủng của vách tim, tâm thất
6.2 Xem thêm
• T - Tracheoesophageal fistula - Rối loạn liên kết
giữa thực quản của hệ thống tiêu hóa và khí quản
của hệ thống hô hấp
• Hội chứng người cá
• Hiện tượng thoái hóa cột sống bẩm sinh
11
12
6.3 Tham khảo và liên kết ngoài
• (tiếng Anh) VACTERL or VATER Association Cincinnati Children’s Hospital Medical Center
• (tiếng Anh) VATER/VACTRL Association University of Kansas Medical Center page with
links to various information and support site
• (tiếng Anh) VACTERL Association Support
Group (UK)
6.4 Tham khảo
CHƯƠNG 6. HỘI CHỨNG VACTERL
Chương 7
Sứt môi và hở hàm ếch
Sứt môi, hở hàm ế và ẻ vòm hầu là các dạng bất
thường bẩm sinh ở môi và miệng thấy khá nhiều trong
các sắc dân Á châu.
7.2 Phân dạng
• 18% sứt môi một bên, không bị chẻ vòm hầu
• 2% sứt môi hai bên, không bị chẻ vòm hầu
• 38% sứt môi một bên và chẻ vòm hầu
• 12% sứt môi hai bên và chẻ vòm hầu
•
• 30% chẻ vòm hầu, không sứt môi. Dạng này tỉ lệ
1/2000, ở nữ nhiều hơn nam. Chia làm nhiều dạng
khác, trong đó có dạng chẻ dưới mô mạc (1/12002000) và dạng chẻ lưỡi gà (1/80).[3]
Sứt môi đôi
7.3 Điều trị
7.3.1 Sơ sinh
•
Sứt môi một bên
Vì các khuyết tật này có thể được chữa trị khi trẻ lớn
trên 1 tuần [4] , mọi cố gắng nên được thực hiện giúp trẻ
sơ sinh có đủ dinh dưỡng để phát triển và đủ sức chịu
đựng cuộc giải phẫu. Các trường hợp nhẹ hơn như sứt
môi, hở hàm ếch phía trước miệng thì trẻ có thể bú mẹ.
Khi trẻ có thể ngậm được cả quầng vú vào miệng thì
một phần vú sẽ che được chỗ hở và trẻ có thể bú tốt
hơn. Trẻ bị chẻ vòm hầu sâu (hở ở thành trên và sau
họng) thì khi bú dễ bị sặc hay trào lên mũi, khó nuốt.
•
Sứt môi một bên, hở hàm Có thể cho uống sữa bằng muỗng hoặc theo ống thông
vào dạ dày.
ếch và chẻ vòm hầu
7.3.2 Phẫu thuật
7.1 Dịch tễ học
Lịch sử
Chứng sứt môi và chẻ vòm hầu đứng đầu trong các
khuyết tật bẩm sinh - khoảng 1/600 trẻ sơ sinh bị chứng
này, từ trường hợp rất nhẹ cho đến nặng.[1] . Tỉ lệ cao
nhất ở dân châu Á, tiếp theo là dân da trắng, và thấp
nhất ở dân da đen. Nam nhiều hơn nữ. Nhiều gia đình
có nhiều con cùng bị khuyết tật - đưa đến giả thuyết có
yếu tố di truyền hay do độc tố môi trường chung quanh
người mẹ. Một số có thêm những khuyết tật bẩm sinh
khác, một số khác lại không.[2]
13
• Nhà hùng biện Hy Lạp Demosthenes (384 – 323
TCN) dùng viên sỏi để lấp lỗ hở vòm hầu để nói
rõ hơn [5]
• Năm 1552 Jacques Houllier đề nghị khâu hai phần
vòm vào nhau.
• Năm 1564 y sĩ tên Pare mô tả cách dùng mô giả
làm lấp khe hở.
14
CHƯƠNG 7. SỨT MÔI VÀ HỞ HÀM ẾCH
• Năm 1764 nha sĩ Pháp LeMonnier thực hiện phẫu 7.3.4
thuật thành công vá chỗ chẻ vòm hầu mềm.
• Năm 1834 Dieffenbach vá cả phần cứng và mềm
của vòm hầu.
• Năm 1861 von Langenbeck sử dụng cách tách mô
bọc xương hai bên kéo vào giữa lấp khe hở
• Năm 1868 Billroth đề nghị cách đập vỡ khúc xương
móc để dễ giải phẫu
• Kỹ thuật của von Langenbeck được các y sĩ Gillies,
Fry, Kilner, Wardill, Veau, và Dorrance lần lượt
phát huy.
• Nhiều bác sĩ và nhà nghiên cứu tranh cãi nhau về
thời gian tốt nhất để giải phẫu. Vì không được giải
quyết thỏa đáng, phẫu thuật vá khe hở bị đình lại.
• Năm 1944 bác sĩ Schweckendiek khởi động và phát
triển lại các cuộc giải phẫu giúp trẻ em bị hở vòm
hầu.[3]
Phát âm
7.4 Chú thích
[1] />[2] />[3] />section~{}introduction
[4] Xu hướng phẫu thuật sớm Tại Bệnh viện Nhi Đồng 1,
đã có những trẻ 1 tuần tuổi được vá sứt môi.
[5] e Cle Palate-Craniofacial Journal: Vol. 37, No. 6, pp.
534–537.
7.5 Liên kết ngoài
(ông tin tiếng Anh về Sứt môi và Hở vòm hầu)
• ư viện Y Khoa ốc gia Hoa Kỳ
• Hội phẫu thuật thẩm mỹ Hoa Kỳ
Tổng quát
Khái niệm chung là nên sửa vòm hầu trước khi trẻ lên
1 tuổi, nghĩa là trước khi nói giỏi. Lợi điểm về khả năng
phát âm quan trọng hơn tác hại của mổ sớm như khi lớn
lên bị hở lại hay làm răng hô, cắn nghiên v.v… Mục đích
của giải phẫu là tạo một nắp ngăn (kín hơi và kín nước)
giữa miệng và mũi, để trẻ có thể nói rõ ràng. Ngoài ra
giải phẫu cũng giúp sự phát triển bình thường của hàm
răng và gương mặt. Ba điều kiện cần thiết của phần
mềm vòm hầu để tạo tiếng nói rõ là phải dài đủ, phải di
động đủ, và phải cong theo hình dạng của vòm khẩu.
Sửa sứt môi có thể thực hiện trước khi sửa vòm hầu và
giúp phát âm tốt hơn. Tuy nhiên có thể sửa môi quá
sớm có thể đưa đến nhiều vấn đề khi trẻ lớn và xương
hàm mặt to lên nhiều hơn.
Kỹ thuật von Langenbeck
Kỹ thuật Schweckendiek
Kỹ thuật Hai phần chắp
Kỹ thuật Wardill-Kilner-Veau
Kỹ thuật vá đôi ngược chữ Z
Kỹ thuật Submucous clefts
Kỹ thuật chắp xương
7.3.3
Nha khoa
Chương 8
Thalidomide
alidomide là một chất màu ngà, không tan trong
nước, tan trong ethanol, hệ số phân tán (partition
coeficient) octanol/nước là 5 ở nhiệt độ trong phòng.
8.1 Công dụng
alidomide ban đầu là một thuốc an thần sử dụng
tương đối rộng rãi tại châu Âu thập kỷ 60 của thế
kỷ 20. alidomide có thể dùng trong điều trị đau
tủy (multiple myeloma) và ban đỏ nốt do bệnh phong
(erythema nodosum leprosum).
8.2 Tác hại đến thai
Sử dụng alidomide trong khi mang thai có thể gây
khiếm khuyết cho cơ thể thai nhi, nhất là làm cụt hai
tay, hai chân. ập kỷ 60 thế kỷ 20 có rất nhiều trường
hợp dị tật thai nhi ở châu Âu do thalidomide. Trong
lịch sử ngành dược gọi đó là “thảm họa thalidomide”.
Sau đó việc thử lâm sàng đối với các thuốc mới rất được
chú trọng.
8.3 Tham khảo
8.4 Liên kết ngoài
• />20090611091540883p0c14/
thalidomide-trong-dieu-tri-benh-da-u-tuy-xuong.
htm
15
Chương 9
Bệnh di truyền
Bệnh di truyền là những bệnh do cha mẹ truyền cho
con qua tế bào sinh dục (trứng hoặc tinh trùng). Vì vậy
mầm bệnh có từ trong hợp tử (phôi), từ điểm khởi thủy
của sự sống trong ổ tử cung. Trên nhiễm sắc thể của
tinh trùng hay trứng đã có sẵn các gen bệnh hoặc cũng
có thể do sai lệch bất thường của nhiễm sắc thể. Có thể
phân loại bệnh di truyền theo chức năng các sản phẩm
của gen bị bệnh: bệnh của phân tử không phải enzim,
bệnh lý của phân tử enzim gây các bệnh về rối loạn
chuyển hoá axít amin, lipit, gluxit… Cũng cần phân biệt
các bệnh di truyền với các bệnh bẩm sinh.
9.1 Tham khảo
16
Chương 10
Bệnh tan máu bẩm sinh
Bệnh thiếu máu vùng biển (alassemia hay biếng ăn. Trẻ sẽ chậm lớn và da hay ửng màu vàng (xem
alassaemia) là tên chung cho một nhóm bệnh thiếu vàng da).
máu.[1]
Nếu không chữa trị, lá lách, gan và tim sẽ sưng to.
Mỗi năm, trên thế giới có khoảng 100.000 em bé sinh Xương bị xốp và dễ gãy, cấu trúc của xương mặt bị
ra mắc phải loại bệnh trầm trọng của nhóm này - phần thay đổi. Vì hồng cầu vỡ sớm hơn bình thường nên tủy
lớn là dân Ý, Hy Lạp, Trung Đông, Nam Á và châu Phi. xương phải làm việc quá sức (để sản xuất hồng cầu),
khiến xương biến dạng. Ở những trẻ bị alassemia
nặng, trán gồ lên, mũi tẹt, xương hàm trên nhô ra do
10.1 Phân loại bệnh Thalassemia tăng sản tủy xương (trẻ em bị thalassemia nặng hay
trông giống nhau vì cấu trúc xương mặt đều bị biến
dạng tương tự). Trẻ sẽ chết sớm, nhất là vì suy tim hay
Nguyên nhân của alassemia là cấu tạo bất bình
nhiễm trùng.
thường của hemoglobin trong hồng cầu. Hai thể dạng
bất thường chính được gọi là alpha-alassemia và beta Trẻ mắc phải dạng thalassemia Trung Đông khi phát
-alassemia, tùy theo phần nào của chất hemoglobin bệnh cũng trầm trọng như trên, nhưng phần đông
bị thiếu. (Hemoglobin là một cấu trúc đạm có khả năng chúng bị nhẹ hơn trong hai thập niên đầu.
giữ dưỡng khí oxygen trong hồng cầu)
Dưới dạng bệnh nặng nhất của alpha-thalassemia, thấy
nhiều nhất trong các sắc dân ở Đông Nam Á, Trung 10.3 Điều trị
ốc và Philippines thường làm hư thai hay trẻ con
chết khi sinh. Phần lớn những người có bệnh alpha- Truyền máu thường xuyên và sử dụng thuốc kháng
thalassemia bị thiếu máu kinh niên - một số bị nặng sinh giúp trẻ bị thalassemia dạng trầm trọng có đời
sống khả quan hơn. Truyền máu thường xuyên (một
trầm trọng, không sống được lâu.
tuần 3-4 lần) có thể làm giảm bớt những biến chứng của
Phần kế tiếp trong bài này liên hệ đến betabệnh (như suy tim hay xốp xương). Nhưng khi truyền
thalassemia.[2] Loại bệnh này cũng bao gồm một số
máu quá nhiều như thế, chất sắt sẽ ứ thừa trong cơ thể,
trường hợp từ trầm trọng cho đến nhẹ, đôi khi không
gây hại cho gan và tim.
có ảnh hường đến sức khỏe:
Hiện nay, khi được chữa trị, người bệnh dạng trầm
trọng thalassemia có thể sống thêm từ 20 đến 30 năm.
1. alassemia dạng trầm trọng, còn được gọi là
bệnh thiếu máu Cooley, theo tên người bác sĩ đầu Một số rất ít có thể chữa bằng phương pháp thay tủy.
tiên miêu tả căn bệnh này vào năm 1925.
2. alassemia dạng trung
3. alassemia dạng nhẹ (hay gọi là “có dấu tích
thalassemia”), thường không có ảnh hưởng đến
sức khỏe, chỉ biết khi thử máu.
10.4 Khả năng di truyền
alassemia là bệnh di truyền dạng lặn của nhiễm sắc
thể thường (tiếng Anh: autosomal recessive). Bệnh này
không có thể lây từ trẻ này sang trẻ khác.
Mỗi con người đều có hai gene (một từ cha và một từ
mẹ) liên hệ đến cấu tạo hemoglobin của hồng cầu. Tạm
gọi gene này là gene 'R' (màu xanh trong thí dụ hình
Khi mới sanh ra, trẻ bị dạng bệnh nặng của thalassemia dưới) và tạm gọi gene có dấu tích thalassemianày là
trông có vẻ khoẻ mạnh bình thường. Nhưng trong vòng gene 'r' (màu đỏ trong thí dụ hình dưới).
vài tháng hay một hai năm, trẻ sẽ khó chịu, mệt mỏi, Ở một người bình thường, cả hai gene đều không có
10.2 Triệu chứng của bệnh
17
18
dấu tích thalassemia (R và R).
CHƯƠNG 10. BỆNH TAN MÁU BẨM SINH
• 3. r (từ cha) - R (từ mẹ) có gene ngầm thalassemia
Một số người có 1 một trong hai gene này có dấu tích
• 4. r (từ cha) - r (từ mẹ) bị thalassemia dạng nặng
thalassemia (R và r). Tuy chỉ có 1 gene bình thường (R),
gene này vẫn có khả năng tạo hemoglobin gần hoàn
hảo, và những người này thường không có triệu chứng Tức là người con có 25% cơ hội hoàn toàn bình thường,
gì cả - chỉ có khi nào họ thử máu mới biết mình có mang 50% có nguy cơ bị mang dấu tích thalassemia và 25% bị
dấu tích bệnh thalassemia.
bệnh thalassemia dạng nặng.
Khi cả hai gene đều bất bình thường, (r và r), thì
hemoglobin sẽ bị cấu tạo sai - gây ra bệnh thalssemia
dạng trầm trọng.
10.5 Thử nghiệm
í dụ các trường hợp di truyền:
• Khám nghiệm máu sẽ cho biết bệnh nhân có bị
thalassemia hay không và thuộc dạng nào.
• Một khi biết có mang dấu tích gene thalassemia,
chồng hay vợ của bệnh nhân cũng nên kiểm tra
máu.
• Khám nghiệm nhau hay nước ối của thai sẽ giúp
bác sĩ biết tính trạng của đứa bé chưa sinh.[3]
Hình 1. Mẹ có gene R-R
Cha có gene R-r
10.6 Phòng ngừa
Bệnh này chưa thể phòng ngừa được, ngoài việc thử
Khi cha có gene ngầm (R - r) nhưng mẹ hoàn toàn bình nghiệm chồng, vợ hay bào thai, giúp giáo dục họ về cơ
thường (R -R) thì người con có thể nằm vào một trong hội xác suất con cái họ có thể mang mầm bệnh hay bị
những trường hợp sau:
bệnh nặng.
• 1. R (từ cha) - R (từ mẹ) không có gene thalassemia
• 2. r (từ cha) - R (từ mẹ) có gene ngầm thalassemia
Tức là người con có 50% nguy cơ bị mang dấu tích
thalassemia, và 50% đứa trẻ sinh ra bình thường.
10.7 Những nghiên cứu đang được
tiến hành
• Tìm cách làm giảm độ ứ thừa của sắt trong cơ thể
của bệnh nhân sau khi được truyền nhiều máu.
Giới khoa học đang tìm và thử các chất có khả
năng thu nhận sắt vào để giúp bài tiềt chất này ra
khỏi cơ thể.
• Điều trị bằng gen
Có thể có những phương hướng sau:
Hình 2. Mẹ có gene R-r.
Cha cũng có gene R-r
Khi cả cha lẫn mẹ đều có gene ngầm (R - r) thì người
con có thể nằm vào một trong những trường hợp sau:
• 1. R (từ cha) - R (từ mẹ) không có gene thalassemia
• 2. R (từ cha) - r (từ mẹ) có gene ngầm thalassemia
1. tìm cách đưa một gene bình thường vào tế bào sơ
khai (stem cells) của bệnh nhân
2. dùng thuốc hay cách gì đó để kích thích những
gene tiếp tục tạo hemoglobin F - loại hemoglobin
này do tủy em bé tạo ra trong khi còn nằm trong
thai nhưng thường bị “tắt” đi khi những loại
hemoglobin khác bắt đầu được cấu tạo.
3. ay tủy
10.9. LIÊN KẾT NGOÀI
10.8 Tham khảo
[1] Mayo Clinic. “alassemia”. Mayo Clinic. Truy cập
ngày 17 tháng 10 năm 2014.
[2] Cianciulli P (tháng 10 năm 2008). “Treatment of iron
overload in thalassemia”. Pediatr Endocrinol Rev 6
(Suppl 1): 208–13. PMID 19337180.
[3] Royal College of Pathologists of Australasia - tiếng Anh
10.9 Liên kết ngoài
• VietNam alassemia Association - Hội tan máu
bẩm sinh Việt Nam, Các thông tin cần biết về bệnh
19
Chương 11
Hội chứng Down
Hội ứng Down là một điều kiện nhiễm sắc thể gây ra
bởi sự hiện diện của tất cả hay một phần của m nhiễm
sắc thể 21 thêm. Tên hội chứng được đặt theo John
Langdon Down, một thầy thuốc đã mô tả hội chứng này
vào năm 1866. Hội chứng này thường gặp nhất trong
số các bệnh do rối loạn nhiễm sắc thể. Cứ 800-1.000 trẻ
mới sinh thì có 1 trẻ bị hội chứng Down.
- ần kinh kém phát triển, bị si đần cao
- Cơ quan sinh dục không phát triển, dẫn đến vô sinh
Ngoài những đặc điểm nói trên, một nửa số trẻ bị Down
có những khuyết tật tim bẩm sinh, song phần lớn có thể
chữa được và sức khỏe của trẻ được cải thiện. Các vấn
đề về hô hấp, tắc nghẽn đường tiêu hóa sớm ở trẻ sơ
sinh và ung thư máu ở tuổi ấu thơ cũng thường gặp. Trẻ
Bình thường, người ta có 46 nhiễm sắc thể (NST), đi bị Down dễ nhạy cảm với các tác nhân nhiễm khuẩn.
thành từng cặp. Một nửa số này được thừa hưởng từ Nhờ những tiến bộ vượt bậc của y học, ngày nay hầu
cha, nửa kia được thừa hưởng từ mẹ. Còn trẻ bị Down hết các vấn đề này đã giải quyết được, do vậy tuổi thọ
lại có 47 NST, nghĩa là có thêm một nhiễm sắc thể 21. trung bình của những người bệnh Down có thể đạt tới
Chính kẻ thừa ra này đã phá vỡ sự phát triển bình 55 tuổi.
thường về thể chất và trí tuệ của trẻ.
Trẻ bị hội chứng Down thường nhỏ hơn những trẻ cùng
Hội chứng có thể xảy ra đối với bất kỳ ai, nhưng nguy trang lứa nhưng lại dễ thừa cân dù theo một chế độ ăn
cơ sẽ cao hơn ở những trẻ sinh ra từ người mẹ ngoài 35 có kiểm soát, nếu tập luyện thường xuyên có thể làm
tuổi. Các thống kê cho thấy, cứ 350 cuộc đẻ của những giảm cân. Trẻ chậm phát triển tâm thần từ thể nhẹ đến
phụ nữ tuổi này có một trẻ sinh ra bị hội chứng Down. thể vừa; nhưng nếu được giúp đỡ và can thiệp kịp thời,
Ở tuổi 40, tỷ lệ này tăng vọt lên 1/100 và tuổi 45 là 1/30.
chỉ gần 10% tiến triển thành thể nặng.
Khoảng 85-90% số thai Down bị chết từ giai đoạn phôi.
Những người sinh ra và sống được phần lớn mắc bệnh Với những trẻ bị hội chứng Down, việc giáo dục kỹ
do sự bất thường ngẫu nhiên trong quá trình thụ tinh, năng thể chất và tâm thần cần được duy trì suốt đời.
và không di truyền. Chỉ có khoảng 5% các trường hợp Nói chung, mức độ chuyển biến trung bình của chúng
thấp hơn những trẻ bình thường; phần lớn dừng lại ở
di truyền.
những kỹ năng vận động, ngôn ngữ và các kỹ năng cá
Trẻ bị Down có nhiều biểu hiện bất thường về hình thái nhân/xã hội đơn giản.
và chức năng:
- Đầu ngắn và bé, gáy rộng và phẳng; cổ ngắn, vai tròn.
11.1 Tham khảo
- Mặt dẹt, trông ngốc.
- Đôi tai thấp nhỏ, dị thường, kém mềm mại.
- Mắt xếch, mí mắt lộn lên, đôi khi bị lác, nếp gấp da
phủ trong mí mắt, mắt hơi sưng và đỏ. Trong lòng đen
có nhiều chấm trắng nhỏ như hạt cát và thường mất đi
sau 12 tháng tuổi.
- Mũi nhỏ và tẹt.
- Miệng trễ và luôn luôn há, vòm miệng cao, lưỡi dày
thè ra ngoài.
- Chân tay ngắn, bàn tay ngắn, to. Các ngón tay ngắn,
ngón út thường khoèo. Lòng bàn tay có nếp sâu nằm
nghiêng. Bàn chân phẳng, ngón chân chim, ngón cái
tòe ra; khoảng cách giữa ngón chân cái và ngón chân
thứ hai quá rộng. Các khớp khuỷu, háng, gối, cổ chân
lỏng lẻo; đôi khi trật khớp háng, trật xương bánh chè.
20
Chương 12
Hội chứng Klinefelter
Hội ứng Klinefelter là tình trạng không phân li
nhiễm sắc thể ở nam giới; người bị tác động có một
cặp nhiễm sắc thể giới tính X thay vì chỉ có một nhiễm
sắc thể X, và có liên hệ với một số nguy cơ về bệnh
lý y học. Hội chứng này được đặt tên theo bác sĩ Harry
Klinefelter, nhà nghiên cứu y học tại Bệnh viên Đa khoa
Massachuses, Boston, Massachuses, do ông lần đầu
tiên miêu tả lâm sàng tình trạng này vào năm 1942.[1]
nhất là 47,XXY (khoảng 80-90% các trường hợp); thể
khảm 46,XY/47,XXY gặp trong khoảng 10% trường hợp.
Các thể khác, gồm 48,XXYY, 48,XXXY, 49,XXXYY, và
49,XXXXY, rất hiếm. ể khảm 46,XY/47,XXY có thể
xảy ra ở hợp tử 46,XY hoặc 47,XXY, và do rối loạn phân
bào sau khi thụ tinh tạo hợp tử.
12.2 Sinh lý bệnh
12.1 Nguyên nhân
Kiểu sắp xếp nhiễm sắc thể XXY là một biến thể di
truyền thường gặp nhất ở kiểu nhân XY, xảy ra vào
khoảng 1 cho mỗi 500 đến 1000 trẻ nam khi sinh ở Hoa
Kỳ, với khoảng 3000 trẻ mới sinh ra mỗi năm. Do sự
thêm vào của một nhiễm sắc thể, người bị tình trạng
này thường được gọi là “Nam XXY”, hay “Nam 47,XXY”
hơn là “mắc hội chứng Klinefelter.”
Sự thêm vào của một hoặc nhiều nhiễm sắc thể X hay
Y lên kiểu nhân nam gây ra các bất thường thực thể và
nhận thức khác nhau. Nói chung, mức độ bất thường
kiểu hình, gồm cả chậm phát triển trí tuệ, liên quan trực
tiếp đến số lượng nhiễm sắc thể X thêm vào trên mức
bình thường. Số nhiễm sắc thể X tăng trên mức bình
thường ảnh hưởng đến phát triển thể chất và nhận thức.
Bất thường xương và tim mạch cũng nặng và tăng hơn.
Phát triển sinh dục đặc biệt nhạy cảm với mỗi nhiễm
sắc thể X thêm vào, gây rối loạn tạo ống sinh tinh và
vô sinh, cũng như dị dạng và thiểu sản cơ quan sinh
dục ở nam đa nhiễm X. Hơn nữa, khả năng tâm thần
giảm theo nhiễm sắc thể X thêm vào. Chỉ số trí tuệ (IQ)
giảm khoảng 15 điểm cho mỗi nhiễm sắc thể X vượt
mức bình thường. Tất cả các khu vực phát triển chính,
gồm ngôn ngữ và sự phối hợp diễn đạt, tiếp thu cũng
bị ảnh hưởng.
Ở động vật có vú với hơn một nhiễm sắc thể X, chỉ có
một nhiễm sắc thể X ở trạng thái hoạt động, còn (các)
nhiễm sắc thể X kia bị bất hoạt và hầu hết các gen trên
chúng không được biểu hiện. Điều này xảy ra cả ở nam
XXY lẫn nữ XX. Tuy nhiên, một số ít gen vẫn được biểu
lộ dù chúng trên nhiễm sắc thể X bất hoạt; đây là các
gen có gen tương ứng trên nhiễm sắc thể Y (vùng giả
nhiễm sắc thể thường). Các gen thể ba nhiễm này ở nam
Hậu quả chính của việc có thêm nhiễm sắc thể giới tính,
XXY có lẽ là nguyên nhân của các triệu chứng liên quan thường mắc phải do lỗi không phân li trong quá trình
đến hội chứng Klinefelter.
tạo giao tử ở cha mẹ, gồm có suy sinh dục, vú to, và
Báo cáo đầu tiên được công bố (Jacobs & Strong 1959)[2] rối loạn tâm lý xã hội. Hội chứng Klinefelter là hình
về người đàn ông có kiểu nhân 47,XXY là bởi Patricia thức suy tinh hoàn nguyên phát, kèm theo tăng mức
A. Jacobs và Bác sĩ J.A. Strong tại Bệnh viện Đa khoa gonadotropin do thiếu phản hồi ngược từ tuyến yên.
Miền Tây ở Edinburgh, Scotland vào năm 1959. Đó là iếu hụt androgen cũng gây tỉ lệ các phần trên cơ thể
một người đàn ông 24 tuổi có biểu hiện của hội chứng giống người bị hoạn; lông ở mặt, nách, mu và thân mình
Klinefelter. Bác sĩ Jacobs miêu tả khám phá của bà về thưa thớt hoặc không có; vú to; tinh hoàn và dương vật
trường hợp dị bội nhiễm sắc thể ở người và động vật có nhỏ; giảm khoái cảm tình dục; phân bố mô mỡ theo
vú đầu tiên được báo cáo trong bài diễn văn cho Giải kiểu phụ nữ; giảm sức chịu đựng thực thể; loãng xương.
thưởng tưởng niệm Wiliam Allan năm 1981 (Jacobs Việc mất ống sinh tinh và tế bào Sertoli dẫn đến giảm
mức inhibin B, chất điều hoà hormone FSH. Trục hạ
1982)[3] .
Trong khoảng 50-60% trường hợp, hiện tượng không đồi-yên-sinh dục bị thay đổi ở bệnh nhân hội chứng
phân li nhiễm sắc thể xảy ra ở mẹ (75% do lỗi phân Klinefelter ở tuổi dậy thì.
bào giảm nhiễm I). Các trường hợp còn lại xảy ra do Cũng có báo cáo về nguy cơ tăng rối loạn tự miễn,
không phân li ở người cha. Kiểu nhân thường gặp như lupus ban đỏ hệ thống, viêm khớp dạng thấp,
21